1. Introduction
Shortly after the entry of human immunodeficiency virus type I (HIV-1) into a target cell, the single-stranded viral RNA genome is reverse-transcribed into a complementary DNA (cDNA) by the virally encoded reverse transcriptase that is transported along with the viral genome in the virus particle (Roberts et al.,
1988). The reverse transcriptase also has ribonuclease H activity that degrades the viral RNA during the synthesis of the cDNA, as well as DNA polymerase activity that creates the sense DNA from the antisense cDNA. Together, the cDNA and its complement form the double-stranded proviral DNA that is then transported into the nucleus, where the proviral DNA is integrated into the host chromosome by the virally encoded integrase and specific host co-factors (Zheng et al.,
2005).
After the HIV-1 provirus is integrated into the host chromosome, it depends on host RNA polymerase II (Pol II) for transcription. The Pol II-mediated transcription can be subdivided into several interconnected stages that include pre-initiation, initiation, promoter clearance, elongation, and termination (Fuda et al.,
2009). During most of the past three decades, major efforts had been devoted to the study of the pre-initiation and initiation stages of the transcription cycle because they were believed to be the principal point where transcription is regulated (Kuras and Struhl,
1999; Ptashne,
2005). In contrast, elongation was considered as an unregulated process that merely involves the repetitive addition of ribonucleotides to the growing RNA chains (Saunders et al.,
2006).
The first hint that this view of transcriptional elongation is questionable or at least not tenable for all genes came from studies of HIV-1, which showed that the elongation phase of Pol II transcription on the integrated proviral DNA is a key rate-limiting step in controlling HIV-1 gene expression (Kao et al.,
1987). Moreover, the independent studies of Bentley and Groudine (
1986) and Rougvie and Lis (
1988) on
c-myc or
hsp70 also identified the promoter proximal pausing as a key regulating step in transcription. A paradigm shift in the transcription field occurred in 2007 on a much larger scale, when global analyses revealed that Pol II pauses at a promoter-proximal region of a large number of genes in both
Drosophila and human stem cells before these genes are activated (Guenther et al.,
2007; Muse et al.,
2007). Subsequent data indicate that the pausing of Pol II soon after initiation is largely due to the actions of negative transcription elongation regulators that include the DRB sensitivity inducing factor (DSIF) and the negative elongation factor (NELF) (Wu et al.,
2003; Yamaguchi et al.,
2013). For the genes containing promoter-proximally paused Pol II, the time-consuming step of setting up a pre-initiation complex has been finished and they are in a state of suspended transcription. The subsequent induction of Pol II elongation can proceed very rapidly and is highly sensitive in response to transcriptional signals. The widespread existence of paused Pol II suggests that elongation plays a much more significant role in regulating gene expression and thus deserves more attention in the field (Guo and Price,
2013).
2. P-TEFb is a key host factor for Tat-dependent HIV-1 transcription
Transcription of the HIV-1 proviral DNA by host Pol II plays a critical role in the viral life cycle because the mRNA is both the template for the synthesis of viral proteins and the genome for progeny viruses. Like all viruses, HIV-1 must replicate by hijacking the cellular machinery to synthesize new virions. However, discrepant with other simpler retroviruses that rely almost entirely on the host, HIV-1 encodes additional accessory proteins that further control the viral life cycle.
As a prime example, the HIV-1-encoded Tat protein is absolutely essential for activating transcriptional elongation from the viral long terminal repeat (LTR) (Jones,
1997). In the absence of Tat, transcription by Pol II is initiated efficiently but generates only short abortive transcripts (Kao et al.,
1987). However, after Tat becomes available, it dramatically enhances the efficiency of Pol II elongation to produce the full-length viral transcripts. Tat accomplishes this task through interacting with the transactivation response (TAR) RNA element, which is a stem-loop structure located at the 5′ end of all nascent viral transcripts and synthesized by Pol II just before pausing.
Almost immediately after the discovery of the Tat-TAR interaction, it became apparently clear that Tat and TAR alone are not sufficient to stimulate HIV-1 transcription. Mounting evidence suggested that specific host cellular cofactors are required in this process (Jones and Peterlin,
1994). After numerous trials and errors, a significant achievement was obtained in the late 1990s with the identification and characterization of the human positive transcription elongation factor b (P-TEFb) as such a cofactor (Marshall and Price,
1995; Marshall et al.,
1996; Mancebo et al.,
1997; Zhu et al.,
1997; Garber et al.,
1998).
P-TEFb is a heterodimer composed of the cyclin-dependent kinase 9 (CDK9) and its regulatory partner cyclin T1 (CycT1). There are also other minor cyclin forms such as cyclin T2a and T2b due to alternative splicing, but they do not interact with Tat (Peng et al.,
1998; Fu et al.,
1999; Ivanov et al.,
1999). P-TEFb is recruited to the HIV-1 LTR through interacting with Tat and TAR. Once positioned next to the paused Pol II, P-TEFb is able to phosphorylate, either directly or indirectly, the C-terminal domain (CTD) of the largest subunit (RPB1) of Pol II on serines at position 2 (Ser2) of the 52 heptapeptide (YSPTSPS) repeats that make up the human Pol II CTD (Fig.
1). Two different kinases are believed to be required for phosphorylation of the CTD (Prelich,
2002). First, CDK7, a component of the multi-subunit general transcription factor TFIIH, phosphorylates serines at position 5 (Ser5) within the heptapeptide repeats. Subsequently, the Ser5-phosphorylated CTD is further phosphorylated by CDK9 at Ser2.
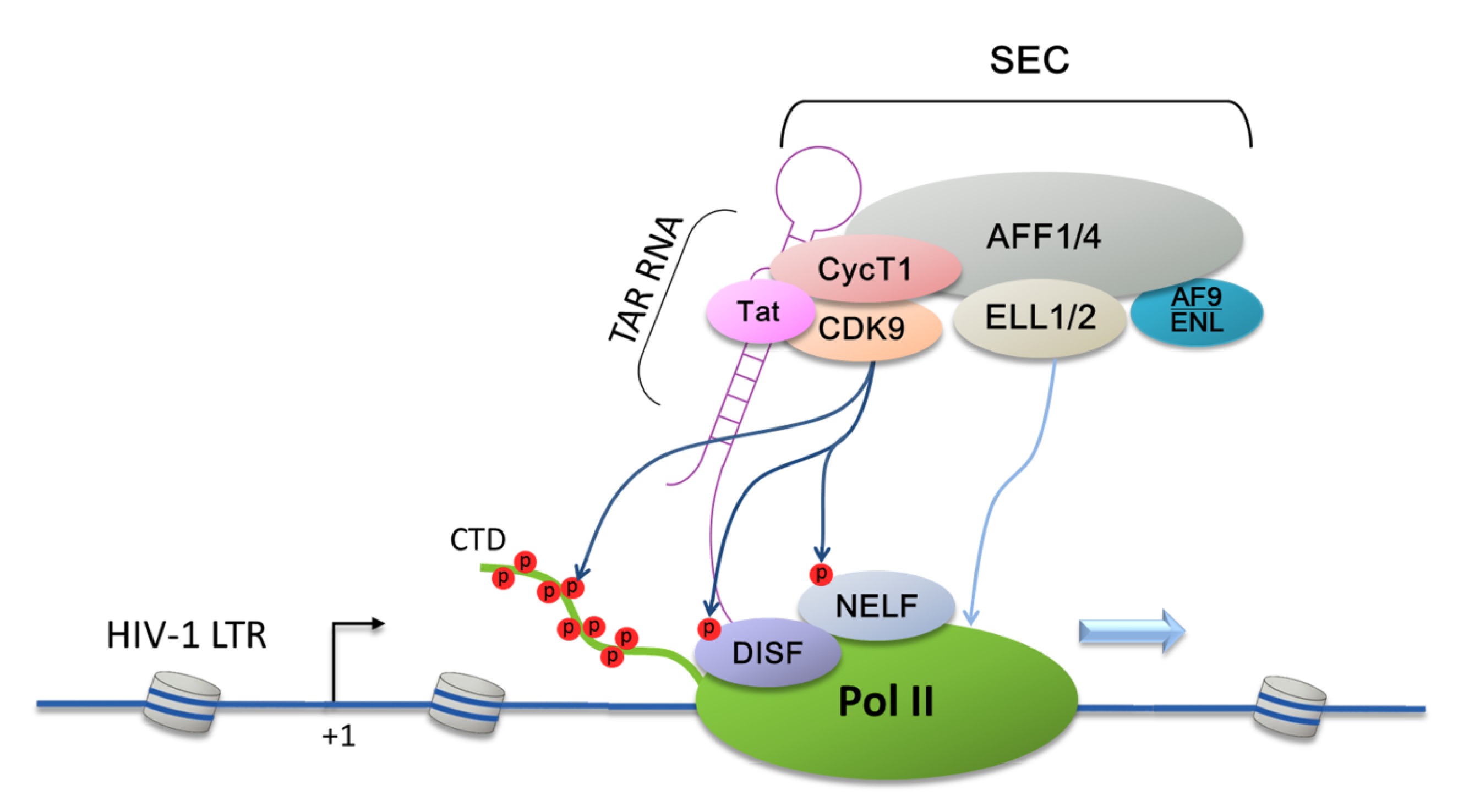
Fig.1
Tat recruitment of super elongation complex (SEC) to the HIV-1 LTR to promote HIV-Tat transactivation
Through interacting with TAR RNA, Tat specifically delivers SEC to the HIV-1 LTR and releases Pol II from promoter-proximal pausing. SEC is a multi-subunit complex that contains P-TEFb, AFF1/4, ELL1/2, and AF9/ENL. Within the SEC, AFF1 or its homolog AFF4 serves as a molecular scaffold, whereas ENL/AF9 uses their N-terminal YEATS domain to target the SEC to Pol II on chromatin. P-TEFb phosphorylates Pol II CTD as well as negative elongation regulators DSIF and NELF. In addition, SEC also contains another well-characterized elongation factor, ELL1/2, which increases the catalytic rate by preventing Pol II backtracking. Thus, Tat significantly activates HIV-1 transcription by recruiting two totally different elongation factors in a single SEC complex
In addition to CTD, both the Spt5 subunit of DSIF (Ivanov et al.,
2000) and the RD subunit of NELF (Fujinaga et al.,
2004) are also substrates for P-TEFb-mediated phosphorylation (Fig.
1). These phosphorylation events antagonize the inhibitory effects of these two negative elongation regulators and release Pol II from promoter-proximal pausing, leading to the production of full-length HIV-1 transcripts.
3. P-TEFb is maintained in a functional equilibrium
P-TEFb is not only required for HIV-1 transcription but also efficient expression of numerous cellular genes (Rahl et al.,
2010). Because of this general dependence on P-TEFb for cellular and viral transcription, the activity of P-TEFb has to be tightly controlled in response to changing cellular requirements for P-TEFb-dependent transcription. It was found that not every CDK9-CycT1 heterodimer in the cell displays the signature P-TEFb kinase and transcriptional activity. Two independent groups subsequently reported that about half of the nuclear P-TEFbs in HeLa cells are sequestered in a kinase-inactive complex that contains the 7SK small nuclear RNA (snRNA) (Nguyen et al.,
2001; Yang et al.,
2001). Produced by RNA Pol III, 7SK is an abundant 331-nucleotide long noncoding RNA that is highly conserved in higher eukaryotes (Wassarman and Steitz,
1991). The 7SK-bound P-TEFb was found to lack the ability to phosphorylate the Pol II CTD on Ser2 and mediate Tat-activation of HIV-1 transcription (Nguyen et al.,
2001; Yang et al.,
2001).
Soon after this discovery, it was found that the 7SK-P-TEFb interaction alone was not enough to inactivate P-TEFb and that the presence of additional factor(s) in the 7SK-P-TEFb complex could be required for the inactivation (Yik et al.,
2003). Indeed, this hypothesis was soon proven to be true by the identification of a nuclear protein called hexamethylene bis-acetamide inducible 1 (HEXIM1) as a P-TEFb/7SK snRNA-associated factor (Michels et al.,
2004; Yik et al.,
2004). It was shown that HEXIM1’s inhibitory effect on P-TEFb is a 7SK snRNA-dependent process that requires the RNA to serve as a molecular scaffold to mediate the interaction between HEXIM1 and P-TEFb.
The identification of novel components of the 7SK snRNA did not just stop at HEXIM1. Subsequent studies reveal that, in addition to HEXIM1, 7SK snRNP also contains the Lupus antigen (La)-related protein 7 (LARP7) and a 7SK snRNA-specific methylphosphate capping enzyme called MePCE (Jeronimo et al.,
2007; He et al.,
2008; Xue et al.,
2010). While MePCE helps protect 7SK snRNA against exonuclease cleavage from the 5′ end (Jeronimo et al.,
2007), the 3′ end of 7SK is occupied by LARP7 that binds to the poly(U) sequence (He et al.,
2008; Markert et al.,
2008). Intriguingly, when bound by LARP7 in the 7SK snRNP, MePCE loses its capping activity and exerts a capping-independent function to promote the LARP7-7SK interaction (Xue et al.,
2010). Thus, the integrity of 7SK snRNP is maintained by the cooperative action of MePCE and LARP7 on 7SK snRNA, which in turn sequesters the excess, unused cellular P-TEFb in an inactive state.
It is interesting to note that although CDK9 lacks kinase activity when sequestered in the 7SK snRNP, it nevertheless has its T-loop already in the phosphorylated state (Chen et al.,
2004; Li et al.,
2005), which is typically associated with an activated CDK with accessible catalytic site (Baumli et al.,
2008). These observations suggest that P-TEFb is only temporarily sequestered in the inactive 7SK snRNP when cellular transcriptional demand is low. Since CDK9 is already phosphorylated on the T-loop and thus poised to become active again, transcription can begin almost immediately once 7SK snRNP is disrupted to release active P-TEFb in response to stimulatory signals (Zhou and Yik,
2006).
Indeed, sequestration of P-TEFb in the 7SK snRNP is completely reversible, and thus the snRNP can be viewed as a major cellular reservoir of unused P-TEFb activity, which can be withdrawn to promote P-TEFb-dependent gene expression (Fig.
2). Consistent with this notion, it has been shown that the exposure of cells to hypertrophic or stress signals, such as flavopiridol, 5,6-dichloro-1-β-D-ribofurano-sylbenzimidazole (DRB), actinomycin D, or UV irradiation, leads to a rapid release of P-TEFb from 7SK snRNP and activation of transcription (Chen et al.,
2004; He et al.,
2006; Biglione et al.,
2007). In addition, activation of TCR pathway in Jurkat T cells by anti-CD3/anti-CD28 antibodies also mediates the disruption of 7SK snRNP and liberation of P-TEFb (Natarajan et al.,
2010). Moreover, the exposure of murine erythroleukemia cells to hexamethylene bisacetamide (HMBA) results in a transient disruption of the complex, which is then followed by a permanent increase in the levels of HEXIM1 and 7SK snRNP after a prolonged treatment (He et al.,
2006).
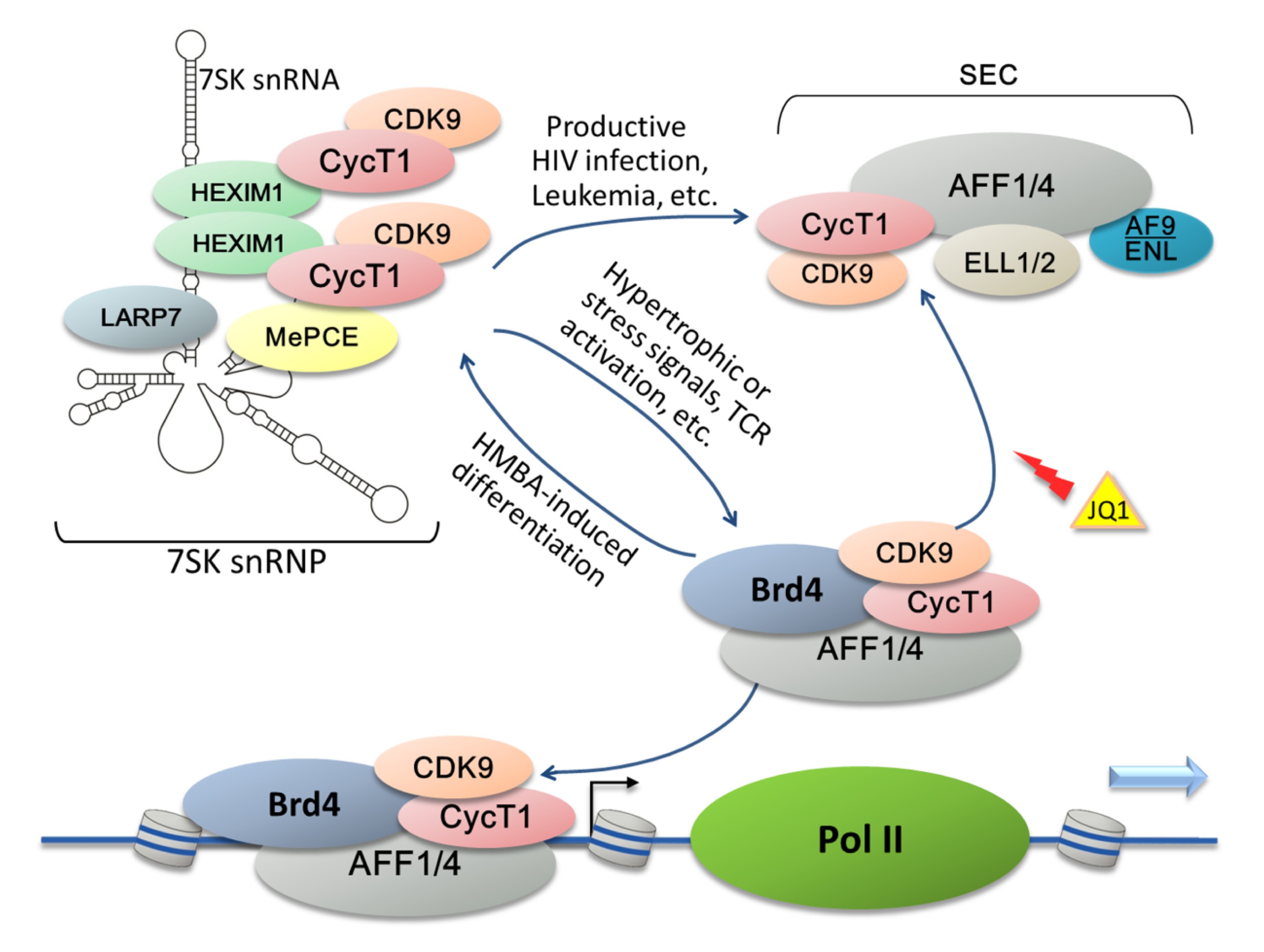
Fig.2
Existence of P-TEFb in different complexes that constitute a dynamic P-TEFb network
P-TEFb is regulated by a reversible association with various transcription factors/cofactors to form several multi-subunit complexes (e.g., 7SK snRNP, super elongation complexes (SECs), and the Brd4-P-TEFb complex) that collectively constitute a P-TEFb network for controlling cellular and HIV transcription. The 7SK snRNP is a cellular reservoir of inactive P-TEFb that requires the 7SK snRNA to serve as a molecular scaffold. The integrity of 7SK snRNP is maintained by LARP7 and MePCE, whereas HEXIM1 inhibits P-TEFb kinase activity. Under specific conditions including TCR activation or the exposure of cells to hypertrophic or stress signals, 7SK snRNP tends to be disrupted to release P-TEFb. Generally, the free P-TEFb is picked up by Brd4 which interacts with acetylated histones and consequently recruits P-TEFb to chromatin to stimulate elongation. Inversely, prolonged treatment with HMBA pushes the P-TEFb equilibrium and leads to reformation of the 7SK snRNP. During HIV-1 infection, Tat competes with Brd4 for binding of free P-TEFb and extracts P-TEFb out of 7SK snRNP to assemble SEC together with several other host factors. The BET bromodomain inhibitor JQ1 efficiently displaces Brd4 from the LTR region of HIV-1 chromatin and increases the association of P-TEFb/SEC with Tat
It is important to point out that there are virtually no free P-TEFb heterodimers in the cell. Once liberated from the 7SK snRNP, most P-TEFbs are found to bind to the bromodomain-containing protein 4 (Brd4), which can interact with the acetylated histones and the transcriptional mediator complex (Jiang et al.,
1998; Dey et al.,
2003). Through the Brd4-P-TEFb interaction, P-TEFb is thus recruited to the promoters of many cellular genes to stimulate transcription (Wu and Chiang,
2007; Dey et al.,
2009). Although the Brd4-P-TEFb interaction appears to be also important for basal HIV-1 transcription, it is found to be inhibitory to Tat-transactivation (Yang et al.,
2005). This is because Brd4 can compete with Tat for binding to the same surface on P-TEFb, which interferes with the more efficient Tat-dependent recruitment of P-TEFb to the HIV-1 promoter (Yang et al.,
2005). Taken together, the published data are consistent with the model that P-TEFb is normally maintained in a functional equilibrium through reversible associations with the positive (Brd4) and negative (HEXIM1/7SK/LARP7/MePCE) regulators (Fig.
2). According to this model, a shift of the P-TEFb equilibrium can produce profound physiological consequences by adjusting the nuclear concentrations of active P-TEFb in accordance to various cellular transcriptional demands and regulatory signals.
4. Tat assembles the super elongation complex (SEC) to promote HIV-1 transcription
It is worth noting that the 7SK-binding motif in HEXIM1 shares high similarity with the arginine-rich TAR-binding motif in the HIV-1 Tat protein (Yik et al.,
2004). In addition, both HEXIM1 and Tat interact with P-TEFb through the same small region near the cyclin box in CycT1 (Karn,
1999; Michels et al.,
2003). It is likely due to these similarities between Tat and HEXIM1 that Tat was found to efficiently displace HEXIM1 and trigger the release of P-TEFb from 7SK snRNP (Barboric et al.,
2007; Sedore et al.,
2007). Consistently, nuclear 7SK snRNP in primary blood lymphocytes shows a marked reduction upon HIV-1 infection (Barboric et al.,
2007).
Upon its extraction of P-TEFb from 7SK snRNP, it is unclear whether Tat assembles only the Tat-TAR-P-TEFb trio on the viral LTR or additional cellular factors may be involved for Tat to fully activate HIV-1 transcription. It is interesting to point out that anecdotal evidence has been reported in the past, which appears to support the latter scenario. For example, the loss of Tat-transactivation in CDK9-depleted nuclear extracts can be rescued by addition of partially purified human P-TEFb, which may contain other associated factors, but not recombinant CDK9-CycT1 (Suñé et al.,
2000). To test the hypothesis that additional factors associated with the CDK9-CycT1 heterodimer of core P-TEFb are also required to promote efficient Tat-transactivation, tandem affinity-purifications were performed to isolate any proteins that may bind to both Flag-tagged CDK9 and HA-tagged Tat in a single complex (He et al.,
2010). This approach has led to the isolation and identification of ELL1/2, AFF1/4, ENL, and AF9 as novel components of the Tat-P-TEFb complex (Fig.
2). Notably, the same set of factors is also independently identified and confirmed through a similar purification scheme that targets Tat only (Sobhian et al.,
2010). This novel multi-subunit complex containing P-TEFb, AFF1/4, ELL1/2, and AF9/ENL is now called SEC.
Within an SEC, AFF1 or its homolog AFF4 serves as a molecular scaffold and uses short interspersed hydrophobic stretches to allow dynamic and flexible assembly of multiple SEC subunits and connect them to each other (He et al.,
2011; Chou et al.,
2013). Although initially thought to be a subunit unique to only the SEC, recent data indicate that AFF1/4 also exists in a portion of 7SK snRNP in the cell (Lu et al.,
2014). Notably, AFF1/4 does not perform a scaffolding role in the 7SK snRNP. Instead, they function to strongly enhance the ability of Tat to extract P-TEFb from 7SK snRNP and form the Tat-SEC complex (Lu et al.,
2014).
Besides AFF1/4, the SEC subunit ENL and its homologous AF9 also make key contributions to SEC’s function. Displaying similar but non-identical functions and existing in separate but closely related SECs, ENL/AF9 uses their N-terminal YEATS (after YNK7, ENL, AF-9, and TFIIF small subunit) domain to target the SEC to Pol II on chromatin through contacting the human PAFc complex, which allows the SEC to stimulate productive elongation (Kim et al.,
2010; He et al.,
2011).
Like P-TEFb, ELL1/2 is also well-characterized transcriptional elongation factors belonging to the ELL family of Pol II elongation-stimulatory factors (Shilatifard et al.,
1997). Employing a different mechanism, ELL1/2 increases the catalytic rate of Pol II by keeping the 3′ end of nascent mRNA in alignment with the catalytic site to prevent Pol II backtracking (Shilatifard et al.,
1996). Thus, two totally different elongation factors, P-TEFb and ELL1/2, are recruited by Tat to the viral LTR in the same SEC complex, where they can work synergistically to release the paused Pol II to stimulate elongation (He et al.,
2010; Lu et al.,
2014) (Fig.
1). This explains why Tat is such a powerful transcriptional activator. Besides HIV-1 Tat, chimeric fusion proteins produced by chromosomal translocations and containing the mixed lineage leukemia (MLL) protein also take advantage of the powerful elongation stimulatory activity of the SEC and recruit the complex to many MLL-target genes to stimulate transcription and cause leukemia (Lin et al.,
2010).
Despite its important role in SEC’s function, ELL2 turns out to be a short-lived protein that undergoes rapid proteasomal degradation (He et al.,
2010). Recently, ELL2 was shown to be a polyubiquitinated protein and the ubiquitination is carried out by the RING (a really interesting new gene) domain protein Siah1, which is identified as an E3 ubiquitin ligase for ELL2 (Liu et al.,
2012). The interaction of ELL2 with the scaffolding protein AFF1/4 dramatically increases the stability of ELL2 and this stabilization is further enhanced by the presence of Tat (He et al.,
2010), which in turn promotes SEC formation and SEC-dependent HIV-1 transcription.
5. Brd4-P-TEFb interaction and its effect on HIV-1 transcription and latency
In cells that are free of HIV-1 infection, Brd4 serves as the cellular equivalent of Tat to recruit P-TEFb to the chromatin loci of many genes, especially the primary response genes, to promote transcriptional elongation (Jang et al.,
2005; Yang et al.,
2005; Delmore et al.,
2011). Brd4 is a ubiquitously expressed nuclear protein belonging to the bromodomain and extraterminal domain (BET) protein family. It contains two N-terminal tandem bromodomains and an extraterminal domain (Jeanmougin et al.,
1997; Dey et al.,
2000). While the two bromodomains are able to interact with acetylated histone H3 and H4 (Dey et al.,
2003), the C terminus of Brd4 is responsible for binding to P-TEFb (Bisgrove et al.,
2007). Thus, P-TEFb can be recruited to the chromatin template by Brd4 and activate gene transcription (Mochizuki et al.,
2008).
Although Brd4 plays a positive role in releasing Pol II from pausing at many cellular gene promoters, it has different effects on Tat-independent and -dependent HIV-1 transcriptions (Jang et al.,
2005; Yang et al.,
2005). While Brd4 elevates the basal Tat-independent HIV-1 transcription, it is a potent inhibitor of Tat-transactivation as it competes with Tat for binding to P-TEFb (Yang et al.,
2005). Furthermore, overexpression of the P-TEFb-interacting domain of Brd4 has been shown to inhibit Tat function and reactivation of HIV-1 latency (Bisgrove et al.,
2007).
As a potent suppressor of Tat-transactivation, it is not surprising that Brd4 has been shown to contribute significantly to the establishment and maintenance of HIV-1 latency (Zhu et al.,
2012; Mbonye et al.,
2013), which is a main obstacle to eradication of infection. Identification of small molecule drugs that can antagonize Brd4’s inhibition of Tat-transactivation and thereby activate latent HIV-1 proviruses may, in the presence of highly active antiretroviral therapy (HAART), permit clearance of infected cells by the immune system (Richman et al.,
2009).
To this end, the BET bromodomain inhibitor JQ1, which binds competitively to the acetyl-lysine recognition motifs or bromodomains in Brd4, has been shown to efficiently reactivate latent HIV-1 in various models (Filippakopoulos et al.,
2010; Banerjee et al.,
2012). JQ1 is able to displace Brd4 from the LTR region of HIV-1 chromatin, thereby antagonizing Brd4’s inhibition of Tat-transactivation (Li et al.,
2013). In-depth analyses reveal that JQ1 significantly increases the association of P-TEFb/SEC with Tat, which results in more SECs recruited to the LTR by Tat and TAR to promote HIV-1 transcriptional elongation (Zhu et al.,
2012; Li et al.,
2013). Besides the inhibition of Brd4-chromatin interaction, JQ1 also leads to a transient release of P-TEFb from the 7SK snRNP. The released P-TEFb was found to associate with Brd4 and the SEC, which also contributes to HIV-1 transcription and latency activation (Bartholomeeusen et al.,
2012; Li et al.,
2013).
It should be noted that JQ1 alone does not appear to reactivate HIV-1 latency very efficiently in infected primary T cells, probably due to the fact that the P-TEFb level in these cells is very low (Chiang and Rice,
2012; Zhu et al.,
2012). However, JQ1 has been shown to cooperate well with other well-known latency activators such as SAHA and HMBA (Bartholomeeusen et al.,
2012), which may potentially prime the system to allow JQ1 to function properly. Finally, since JQ1 has been developed as an anti-cancer and growth-suppressive agent due to its inhibition of Brd4-dependent recruitment of P-TEFb to many primary response genes such as
c-myc (Delmore et al.,
2011; Bartholomeeusen et al.,
2012), its Tat-specific effect in activating HIV-1 latency ensures that it does not induce general T cell activation, an unwanted side effect of latency reactivation. Indeed, the changes in global gene expression induced by JQ1 or αCD3/αCD28 have been compared through a microarray analysis, which indicates that JQ1 down-regulates T cell activation genes but up-regulates histone modification genes (Banerjee et al.,
2012).
Besides inhibition of Tat-transactivation, several other mechanisms are also associated with the establishment of HIV-1 latency, which include transcriptional interference, limited availability of transcription factors, chromatin modifications, DNA methylation, etc. Since we are focusing on P-TEFb and related factors, other molecular mechanisms of HIV-1 latency will not be discussed in this review. For more details of HIV-1 latency, please refer to relevant recent reviews (Donahue and Wainberg,
2013; Ruelas and Greene,
2013).
6. Conclusions and perspectives
More than thirty years have passed since HIV-1 was first identified as the causative agent of acquired immune deficiency syndrome (AIDS). Significant progress has been made toward finding effective ways to suppress viral replication. The current antiretroviral therapy (ART) can inhibit the activities of several viral enzymes, prevent virus replication, and protect the immune system of the host. However, life-long treatment is still compulsive because of the existence of long-lived latency reservoirs, and there are serious side effects associated with the therapy (Hakre et al.,
2012). To eradicate the latent reservoirs, the so-called “shock and kill” strategy has been proposed, which combines the HIV-1 latency reactivation and ART treatment to achieve a functional cure for HIV/AIDS (Richman et al.,
2009; Karn,
2011; Siliciano and Greene,
2011; Archin et al.,
2012).
Towards this goal, high specific and efficient HIV-1 latency activators have to be developed first. Since P-TEFb, a very important human transcription factor and Tat cofactor, is not present on the HIV-1 LTR in latent cells, devising effective ways to release P-TEFb from the 7SK snRNP and promote the formation of the Tat-SEC complex on the viral promoter offers a promising strategy to activate HIV-1 transcription and latency. Furthermore, molecular insights into the structures and functions of the Tat-SEC complex and 7SK snRNP may reveal novel targets for developing more specific and effective antiviral drugs. Although there is still a very long and hard way to go before we can fully cure HIV/AIDS, fortunately we are already on the road and heading in the right direction. In addition, through a better understanding of the molecular mechanism that controls HIV-1 transcription and latency, a cure for HIV/AIDS is no longer an impossible task in our lifetime.
* Project supported by the National Natural Science Foundation of China (No. 81201276) and the Natural Science Foundation of Fujian Province (No. 2012J05067), ChinaCompliance with ethics guidelines Rong-diao LIU, Jun WU, Rui SHAO, and Yu-hua XUE declare that they have no conflict of interest.References
[1] Archin, N.M., Liberty, A.L., Kashuba, A.D., 2012. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy.
Nature, 487(7408):482-485.


[2] Banerjee, C., Archin, N., Michaels, D., 2012. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1.
J Leukoc Biol, 92(6):1147-1154.
[3] Barboric, M., Yik, J.H., Czudnochowski, N., 2007. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription.
Nucl Acids Res, 35(6):2003-2012.
[4] Bartholomeeusen, K., Xiang, Y., Fujinaga, K., 2012. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein.
J Biol Chem, 287(43):36609-36616.
[5] Baumli, S., Lolli, G., Lowe, E.D., 2008. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation.
EMBO J, 27(13):1907-1918.
[6] Bentley, D.L., Groudine, M., 1986. Novel promoter upstream of the human
c-myc gene and regulation of
c-myc expression in B-cell lymphomas.
Mol Cell Biol, 6(10):3481-3489.
[7] Biglione, S., Byers, S.A., Price, J.P., 2007. Inhibition of HIV-1 replication by P-TEFb inhibitors DRB, seliciclib and flavopiridol correlates with release of free P-TEFb from the large, inactive form of the complex.
Retrovirology, 4(1):47
[8] Bisgrove, D.A., Mahmoudi, T., Henklein, P., 2007. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription.
PNAS, 104(34):13690-13695.
[9] Chen, R., Yang, Z., Zhou, Q., 2004. Phosphorylated positive transcription elongation factor b (P-TEFb) is tagged for inhibition through association with 7SK snRNA.
J Biol Chem, 279(6):4153-4160.
[10] Chiang, K., Rice, A.P., 2012. MicroRNA-mediated restriction of HIV-1 in resting CD4
+ T cells and monocytes.
Viruses, 4(12):1390-1409.
[11] Chou, S., Upton, H., Bao, K., 2013. HIV-1 Tat recruits transcription elongation factors dispersed along a flexible AFF4 scaffold.
PNAS, 110(2):E123-E131.
[12] Delmore, J.E., Issa, G.C., Lemieux, M.E., 2011. BET bromodomain inhibition as a therapeutic strategy to target c-Myc.
Cell, 146(6):904-917.
[13] Dey, A., Ellenberg, J., Farina, A., 2000. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G
2-to-M transition.
Mol Cell Biol, 20(17):6537-6549.
[14] Dey, A., Chitsaz, F., Abbasi, A., 2003. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis.
PNAS, 100(15):8758-8763.
[15] Dey, A., Nishiyama, A., Karpova, T., 2009. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription.
Mol Biol Cell, 20(23):4899-4909.
[16] Donahue, D.A., Wainberg, M.A., 2013. Cellular and molecular mechanisms involved in the establishment of HIV-1 latency.
Retrovirology, 10(1):11
[17] Filippakopoulos, P., Qi, J., Picaud, S., 2010. Selective inhibition of BET bromodomains.
Nature, 468(7327):1067-1073.
[18] Fu, T.J., Peng, J., Lee, G., 1999. Cyclin K functions as a CDK9 regulatory subunit and participates in RNA polymerase II transcription.
J Biol Chem, 274(49):34527-34530.
[19] Fuda, N.J., Ardehali, M.B., Lis, J.T., 2009. Defining mechanisms that regulate RNA polymerase II transcription
in vivo
.
Nature, 461(7261):186-192.
[20] Fujinaga, K., Irwin, D., Huang, Y., 2004. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element.
Mol Cell Biol, 24(2):787-795.
[21] Garber, M.E., Wei, P., Jones, K.A., 1998. HIV-1 Tat interacts with cyclin T1 to direct the P-TEFb CTD kinase complex to TAR RNA.
Cold Spring Harb Symp Quant Biol, 63:371-380.
[22] Guenther, M.G., Levine, S.S., Boyer, L.A., 2007. A chromatin landmark and transcription initiation at most promoters in human cells.
Cell, 130(1):77-88.
[23] Guo, J., Price, D.H., 2013. RNA polymerase II transcription elongation control.
Chem Rev, 113(11):8583-8603.
[24] Hakre, S., Chavez, L., Shirakawa, K., 2012. HIV latency: experimental systems and molecular models.
FEMS Microbiol Rev, 36(3):706-716.
[25] He, N., Pezda, A.C., Zhou, Q., 2006. Modulation of a P-TEFb functional equilibrium for the global control of cell growth and differentiation.
Mol Cell Biol, 26(19):7068-7076.
[26] He, N., Jahchan, N.S., Hong, E., 2008. A La-related protein modulates 7SK snRNP integrity to suppress P-TEFb-dependent transcriptional elongation and tumorigenesis.
Mol Cell, 29(5):588-599.
[27] He, N., Liu, M., Hsu, J., 2010. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription.
Mol Cell, 38(3):428-438.
[28] He, N., Chan, C.K., Sobhian, B., 2011. Human polymerase-associated factor complex (PAFc) connects the super elongation complex (SEC) to RNA polymerase II on chromatin.
PNAS, 108(36):E636-E645.
[29] Ivanov, D., Kwak, Y.T., Nee, E., 1999. Cyclin T1 domains involved in complex formation with Tat and TAR RNA are critical for
tat-activation.
J Mol Biol, 288(1):41-56.
[30] Ivanov, D., Kwak, Y.T., Guo, J., 2000. Domains in the SPT5 protein that modulate its transcriptional regulatory properties.
Mol Cell Biol, 20(9):2970-2983.
[31] Jang, M.K., Mochizuki, K., Zhou, M., 2005. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription.
Mol Cell, 19(4):523-534.
[32] Jeanmougin, F., Wurtz, J.M., Le Douarin, B., 1997. The bromodomain revisited.
Trends Biochem Sci, 22(5):151-153.
[33] Jeronimo, C., Forget, D., Bouchard, A., 2007. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme.
Mol Cell, 27(2):262-274.
[34] Jiang, Y.W., Veschambre, P., Erdjument-Bromage, H., 1998. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways.
PNAS, 95(15):8538-8543.
[35] Jones, K.A., 1997. Taking a new TAK on tat transactivation.
Genes Dev, 11(20):2593-2599.
[36] Jones, K.A., Peterlin, B.M., 1994. Control of RNA initiation and elongation at the HIV-1 promoter.
Annu Rev Biochem, 63(1):717-743.
[37] Kao, S.Y., Calman, A.F., Luciw, P.A., 1987. Anti-termination of transcription within the long terminal repeat of HIV-1 by
tat gene product.
Nature, 330(6147):489-493.
[38] Karn, J., 1999. Tackling tat.
J Mol Biol, 293(2):235-254.
[39] Karn, J., 2011. The molecular biology of HIV latency: breaking and restoring the Tat-dependent transcriptional circuit.
Curr Opin HIV AIDS, 6(1):4-11.
[40] Kim, J., Guermah, M., Roeder, R.G., 2010. The human PAF1 complex acts in chromatin transcription elongation both independently and cooperatively with SII/TFIIS.
Cell, 140(4):491-503.
[41] Kuras, L., Struhl, K., 1999. Binding of TBP to promoters
in vivo is stimulated by activators and requires Pol II holoenzyme.
Nature, 399(6736):609-613.
[42] Li, Q., Price, J.P., Byers, S.A., 2005. Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186.
J Biol Chem, 280(31):28819-28826.
[43] Li, Z., Guo, J., Wu, Y., 2013. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation.
Nucl Acids Res, 41(1):277-287.
[44] Lin, C., Smith, E.R., Takahashi, H., 2010. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia.
Mol Cell, 37(3):429-437.
[45] Liu, M., Hsu, J., Chan, C., 2012. The ubiquitin ligase Siah1 controls ELL2 stability and formation of super elongation complexes to modulate gene transcription.
Mol Cell, 46(3):325-334.
[46] Lu, H., Li, Z., Xue, Y., 2014. AFF1 is a ubiquitous P-TEFb partner to enable Tat extraction of P-TEFb from 7SK snRNP and formation of SECs for HIV transactivation.
PNAS, 111(1):E15-E24.
[47] Mancebo, H.S., Lee, G., Flygare, J., 1997. P-TEFb kinase is required for HIV Tat transcriptional activation
in vivo and
in vitro
.
Genes Dev, 11(20):2633-2644.
[48] Markert, A., Grimm, M., Martinez, J., 2008. The La-related protein LARP7 is a component of the 7SK ribonucleoprotein and affects transcription of cellular and viral polymerase II genes.
EMBO Rep, 9(6):569-575.
[49] Marshall, N.F., Price, D.H., 1995. Purification of P-TEFb, a transcription factor required for the transition into productive elongation.
J Biol Chem, 270(21):12335-12338.
[50] Marshall, N.F., Peng, J., Xie, Z., 1996. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase.
J Biol Chem, 271(43):27176-27183.
[51] Mbonye, U.R., Gokulrangan, G., Datt, M., 2013. Phosphorylation of CDK9 at Ser175 enhances HIV transcription and is a marker of activated P-TEFb in CD4
+ T lymphocytes.
PLoS Pathog, 9(5):e1003338
[52] Michels, A.A., Nguyen, V.T., Fraldi, A., 2003. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner.
Mol Cell Biol, 23(14):4859-4869.
[53] Michels, A.A., Fraldi, A., Li, Q., 2004. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor.
EMBO J, 23(13):2608-2619.
[54] Mochizuki, K., Nishiyama, A., Jang, M.K., 2008. The bromodomain protein Brd4 stimulates G
1 gene transcription and promotes progression to S phase.
J Biol Chem, 283(14):9040-9048.
[55] Muse, G.W., Gilchrist, D.A., Nechaev, S., 2007. RNA polymerase is poised for activation across the genome.
Nat Genet, 39(12):1507-1511.
[56] Natarajan, M., August, A., Henderson, A.J., 2010. Combinatorial signals from CD28 differentially regulate human immunodeficiency virus transcription in T cells.
J Biol Chem, 285(23):17338-17347.
[57] Nguyen, V.T., Kiss, T., Michels, A.A., 2001. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes.
Nature, 414(6861):322-325.
[58] Peng, J., Zhu, Y., Milton, J.T., 1998. Identification of multiple cyclin subunits of human P-TEFb.
Genes Dev, 12(5):755-762.
[59] Prelich, G., 2002. RNA polymerase II carboxy-terminal domain kinases: emerging clues to their function.
Eukaryot Cell, 1(2):153-162.
[60] Ptashne, M., 2005. Regulation of transcription: from lambda to eukaryotes.
Trends Biochem Sci, 30(6):275-279.
[61] Rahl, P.B., Lin, C.Y., Seila, A.C., 2010. c-Myc regulates transcriptional pause release.
Cell, 141(3):432-445.
[62] Richman, D.D., Margolis, D.M., Delaney, M., 2009. The challenge of finding a cure for HIV infection.
Science, 323(5919):1304-1307.
[63] Roberts, J.D., Bebenek, K., Kunkel, T.A., 1988. The accuracy of reverse transcriptase from HIV-1.
Science, 242(4882):1171-1173.
[64] Rougvie, A.E., Lis, J.T., 1988. The RNA polymerase II molecule at the 5′ end of the uninduced
hsp70 gene of
D. melanogaster is transcriptionally engaged.
Cell, 54(6):795-804.
[65] Ruelas, D.S., Greene, W.C., 2013. An integrated overview of HIV-1 latency.
Cell, 155(3):519-529.
[66] Saunders, A., Core, L.J., Lis, J.T., 2006. Breaking barriers to transcription elongation.
Nat Rev Mol Cell Biol, 7(8):557-567.
[67] Sedore, S.C., Byers, S.A., Biglione, S., 2007. Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR.
Nucl Acids Res, 35(13):4347-4358.
[68] Shilatifard, A., Lane, W.S., Jackson, K.W., 1996. An RNA polymerase II elongation factor encoded by the human
ELL gene.
Science, 271(5257):1873-1876.
[69] Shilatifard, A., Duan, D.R., Haque, D., 1997. ELL2, a new member of an ELL family of RNA polymerase II elongation factors.
PNAS, 94(8):3639-3643.
[70] Siliciano, R.F., Greene, W.C., 2011. HIV latency.
Cold Spring Harb Perspect Med, 1(1):a007096
[71] Sobhian, B., Laguette, N., Yatim, A., 2010. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP.
Mol Cell, 38(3):439-451.
[72] Suñ, C., Goldstrohm, A.C., Peng, J., 2000. An
in vitro transcription system that recapitulates equine infectious anemia virus Tat-mediated inhibition of human immunodeficiency virus type 1 Tat activity demonstrates a role for positive transcription elongation factor b and associated proteins in the mechanism of Tat activation.
Virology, 274(2):356-366.
[73] Wassarman, D.A., Steitz, J.A., 1991. Structural analyses of the 7SK ribonucleoprotein (RNP), the most abundant human small RNP of unknown function.
Mol Cell Biol, 11(7):3432-3445.
[74] Wu, C.H., Yamaguchi, Y., Benjamin, L.R., 2003. NELF and DSIF cause promoter proximal pausing on the
hsp70 promoter in
Drosophila
.
Genes Dev, 17(11):1402-1414.
[75] Wu, S.Y., Chiang, C.M., 2007. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation.
J Biol Chem, 282(18):13141-13145.
[76] Xue, Y., Yang, Z., Chen, R., 2010. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP.
Nucl Acids Res, 38(2):360-369.
[77] Yamaguchi, Y., Shibata, H., Handa, H., 2013. Transcription elongation factors DSIF and NELF: promoter-proximal pausing and beyond.
Biochim Biophys Acta, 1829(1):98-104.
[78] Yang, Z., Zhu, Q., Luo, K., 2001. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription.
Nature, 414(6861):317-322.
[79] Yang, Z., Yik, J.H., Chen, R., 2005. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4.
Mol Cell, 19(4):535-545.
[80] Yik, J.H., Chen, R., Nishimura, R., 2003. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA.
Mol Cell, 12(4):971-982.
[81] Yik, J.H., Chen, R., Pezda, A.C., 2004. A human immunodeficiency virus type 1 Tat-like arginine-rich RNA-binding domain is essential for HEXIM1 to inhibit RNA polymerase II transcription through 7SK snRNA-mediated inactivation of P-TEFb.
Mol Cell Biol, 24(12):5094-5105.
[82] Zheng, Y.H., Lovsin, N., Peterlin, B.M., 2005. Newly identified host factors modulate HIV replication.
Immunol Lett, 97(2):225-234.
[83] Zhou, Q., Yik, J.H., 2006. The Yin and Yang of P-TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation.
Microbiol Mol Biol Rev, 70(3):646-659.
[84] Zhu, J., Gaiha, G.D., John, S.P., 2012. Reactivation of latent HIV-1 by inhibition of BRD4.
Cell Rep, 2(4):807-816.
[85] Zhu, Y., Peery, T., Peng, J., 1997. Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation
in vitro
.
Genes Dev, 11(20):2622-2632.
Open peer comments: Debate/Discuss/Question/Opinion
<1>