1. Introduction
Prostate cancer (PCa) is the second most common noncutaneous cancer in men (exceeded only by lung cancer), accounting for 14% of all new cancer cases in men worldwide. In 2008, 900 000 cases were identified, and 258 000 of them resulted in death (Jemal et al.,
2011). These dark figures resulted in the identification of PCa as the sixth leading cause of death from all cancers among men worldwide (Ferlay et al.,
2010).
Old age, black ethnicity, and a family history of the disease are the risk factors most commonly associated with PCa. The average age at the time of diagnosis is 67 years and about two-thirds of cases are diagnosed in men aged 65 years and over-diagnosis before age 40 years is rare. Environmental risk factors such as eating habits, early sexual initiation, and sexually transmitted infections, both viral (herpes simplex virus 2, human papillomaviruses 18 and 16, and human cytomegalovirus) and bacterial (
Neisseria gonorrhoea,
Treponema pallidum, and
Chlamydia trachomatis), are also associated with the disease (Nelson et al.,
2003).
In terms of geographic variation, developed countries account for about 72% of all diagnosed PCa cases and 53% of all deaths related to the condition. PCa incidence is high in Australia and New Zealand, whereas South-Central Asia has the lowest incidence rate. Variation in PCa incidence is partly attributed to differences in access to diagnostic and treatment procedures (Ferlay et al.,
2010; Jemal et al.,
2011).
Interestingly, as disease incidence has been increasing, mortality has been decreasing markedly. This is due, in part, to the early detection of the disease as a result of the widespread implementation of prostate-specific antigen (PSA) screening, and significant advances in PCa therapeutics (Miller et al.,
2003). Nonetheless, the use of PSA screening is controversial because the early detection of PCa based on this measurement can lead to the diagnosis and treatment of PCa cases that would not otherwise cause symptoms or threaten life (Schröder et al.,
2006; Lumen et al.,
2012). Recently published data suggest that PSA-based screening results in the detection of more cases of PCa, but the effects on PCa mortality are minimal or non-existent (Djulbegovic et al.,
2010; Ilic et al.,
2011). These findings are aggravated when considering that PCa treatment can have major side effects, including urinary incontinence and sexual impotence.
On the other hand, advances in PCa treatment are recognized as a crucial factor in reducing PCa mortality. For instance, sipuleucel T, a cancer treatment vaccine approved by the US Food and Drug Administration (FDA) in 2010, improves the survival of men with advanced PCa (Kantoff et al.,
2010; Thara et al.,
2011). However, advanced hormone resistant or metastatic disease continues to challenge medical management, which can offer only palliative care in most of these cases. Therefore, effective treatment and management of advanced PCa is still an important preoccupation in clinical practice (Turner et al.,
2011; Drudge-Coates and Turner,
2012).
The identification of new molecular markers not only allows a more reliable prediction of the pathological stage of the disease, but can also be useful in the process of selecting therapeutic targets: the ideal scenario would be a panel of molecular biomarkers.
In this review, we briefly describe the pathophysiological features of PCa, focusing on the main molecular processes involved. We also analyze the conventional approaches to PCa diagnosis and treatment. Lastly, we explore recent advances in the establishment of new molecular markers and therapeutic targets for PCa management.
2. Biology of prostate cancer
2.1. Prostate cancer precursors
Despite controversies concerning precancerous prostate lesions, proliferative inflammatory atrophy (PIA), prostatic intraepithelial neoplasia (PIN), and atypical small acinar proliferation (ASAP) have been accepted as the main precursors of PCa (Bostwick et al.,
1996; de Marzo et al.,
1999; Putzi and de Marzo,
2000; Samaratunga et al.,
2006; Davidsson et al.,
2011). Several mechanisms have been identified as triggers for each stage of progression (Fig.
1).
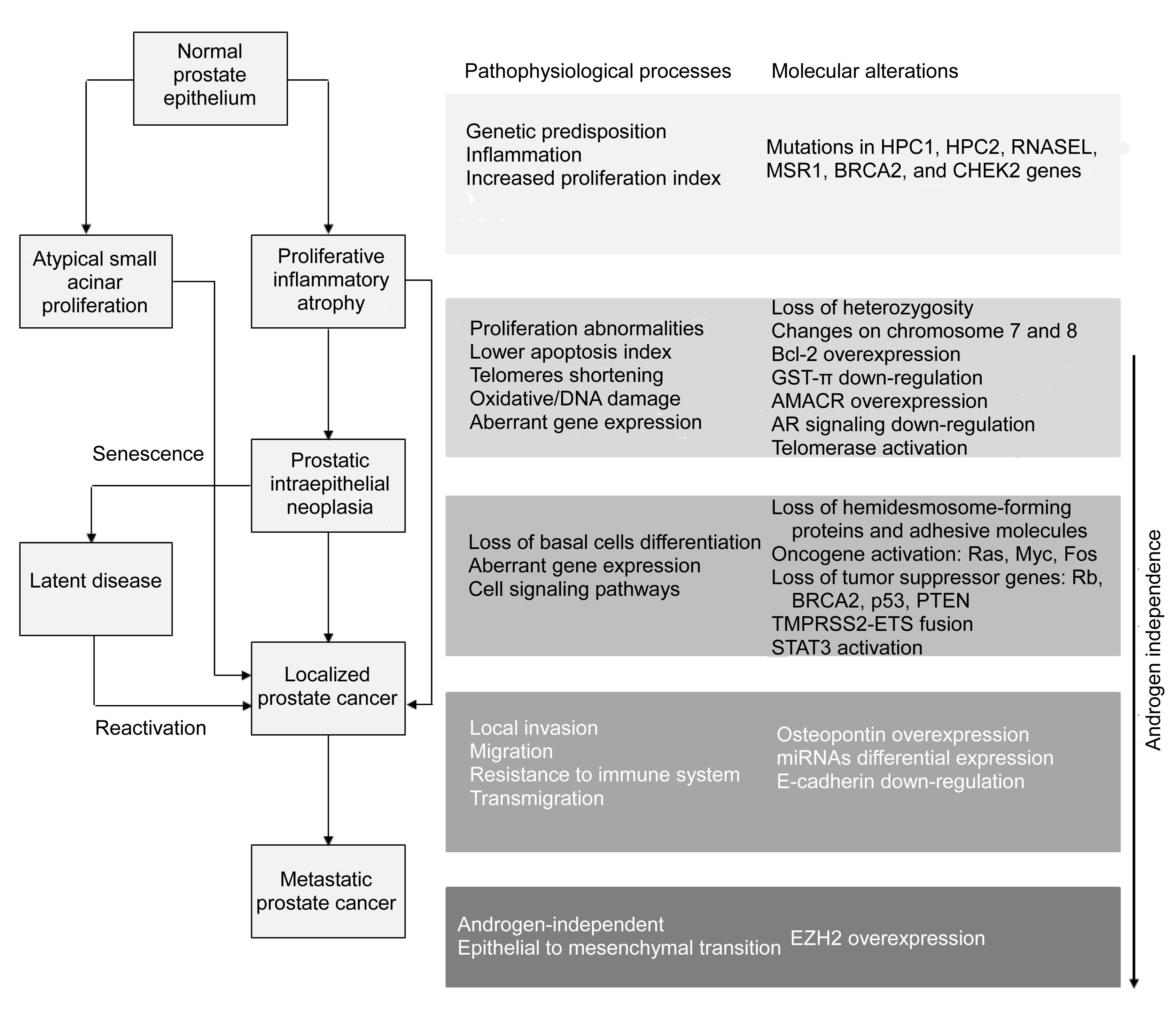
Fig.1
Stages and processes underlying human prostate cancer (PCa) initiation and development
Genetic predisposition, inflammation, and increased cell proliferation are determinant factors for PCa initiation. The occurrence of these processes in the normal prostate epithelium gives rise to proliferative inflammatory atrophy (PIA) or atypical small acinar proliferation (ASAP). These lesions can directly progress to primary PCa. PIA might also evolve to an intermediate stage, prostatic intraepithelial neoplasia (PIN), in which a loss of proliferation capability in basal cell layers coincides with an increase in the proliferation activity of luminal secretory cells. Loss of tumor suppressor genes, oncogene activation, and altered cell signaling promote the progression to localized PCa. For instance, the loss of PTEN in addition to TMPRSS2-ETS fusion (mainly with the v-ets erythroblastosis virus E26 oncogene homolog (avian) member of the ETS family, ERG) promotes progression to PCa through downstream pathways. Alternatively, cells in PIN may undergo a senescence process in which the disease enters a latent stage which, nonetheless, can be reactivated. The establishment of metastases encompasses alterations in cell-cell and cell-matrix interactions that culminate in local invasion, migration, survival to the immune system, and transmigration to secondary sites. While PCa progresses, the dependence on androgens decreases until a completely androgen-independent cancer is formed. The scheme on the left represents the stages of PCa progression up until a metastatic and androgen-independent stage. On the right, each grey rectangle relates to the phase of progression shown immediately to the left. AMACR, α-methylacyl-CoA racemase; AR, androgen receptor; BRCA2, breast cancer 2, early onset; CHEK2, checkpoint kinase 2; ETS, E-twenty six; GST-π, glutathione-S-transferase π; HPC1, hereditary prostate cancer 1; HPC2, hereditary prostate cancer 2; miRNAs, microRNAs; MSR1, macrophage scavenger receptor 1 gene; p53, protein 53; PTEN, phosphatase and tensin homologue; Rb, retinoblastoma tumor suppressor gene; RNASEL, ribonuclease L (2′,5′-oligoisoadenylate synthetase-dependent); STAT3, signal transducer and activator of transcription 3; TMPRSS2, transmembrane protease serine 2
PIA consists of a chronic inflammation found mainly in the peripheral zone of the prostate. The hallmarks of PIA are focal glandular atrophy, a low frequency of apoptosis, and a high proliferation of epithelial cells. Apparently, atrophic cells in PIA undergo malignant transformation and originate PCa either directly or indirectly via development of high-grade PIN (HGPIN). This mechanism is corroborated by the common presence of PIA near foci of HGPIN or PCa (Putzi and de Marzo,
2000; Wang et al.,
2009).
Like PIA, PIN is found primarily in the peripheral zone (Bostwick and Brawer,
1987). PIN presents many of the genotypic alterations, architectural and cytological features, and markers of differentiation of early invasive carcinoma (Bostwick and Brawer,
1987; Bostwick et al.,
1996; Abate-Shen and Shen,
2000). However, unlike PIA, the basement membrane remains intact in PIN (avoiding stromal invasion) and cells express the same proteins and integrin receptors as in normal or hyperplastic stages (Bonkhoff,
1998). Also, these lesions do not produce high levels of PSA (Abate-Shen and Shen,
2000). Microscopically, changes observed in PIN are a continuum between low-grade (almost normal) and high-grade (abnormal) forms thought to be the immediate precursors of early invasive carcinoma (Abate-Shen and Shen,
2000). Alternatively, PIN cells might undergo a senescence period in which their viability is preserved but no proliferation is observed. This phenotype was experimentally demonstrated in cells with complete inactivation of the phosphatase and tensin homolog (PTEN). It was enhanced in the presence of S-phase kinase-associated protein 2 (Skp2) E3-ubiquitin ligase inactivation, and reversed with the inactivation of p53 (Chen et al.,
2005; Lin et al.,
2010).
ASAP is the precancerous lesion most related to PCa (Moore et al.,
2005; Amin et al.,
2007; Lopez,
2007). By definition, ASAP consists of a focus of small acinar structures formed by atypical epithelial cells and is considered a strong risk factor for PCa. Consequently, a follow-up of patients with ASAP is mandatory (Mancuso et al.,
2007; Koca et al.,
2011).
2.2. Underlying prostate cancer development and metastasis formation
PCa is a relatively heterogeneous and multifocal disorder. Around 70% of PCa cases originate in the peripheral zone, 15%–20% in the central zone, and only 10%–15% in the transition zone of the prostate gland (Crawford,
2009). PCa can assume diverse histological patterns, but most cases correspond to acinar adenocarcinomas that arise from prostatic epithelial cells which express androgen receptor (AR) (Bostwick,
1989). Ductal adenocarcinomas, mucinous carcinomas, and signet ring adenocarcinomas are extremely rare (Grignon,
2004).
The cellular origin of PCa has been attributed either to the acquisition of dedifferentiation and mortality by differentiated luminal cells or to the malignant transformation of prostate stem cells that reside among the basal cells (Knudsen and Vasioukhin,
2010). It has also been observed that PCa can arise from basal cells, although the aggressive potential of luminal and basal cell populations differs (Lu et al.,
2013).
The severe differentiation and proliferation abnormalities that underlie PCa development might involve multiple genetic changes, such as loss of heterozygosity, activation of oncogenes, and loss of tumor suppressor genes (Foster et al.,
2000). Several other factors have been associated with PCa development and progression (Fig.
1). Most are a consequence of the natural aging process—age is the most significant risk factor for PCa development. In addition to the genetic causes already mentioned, factors that contribute to the carcinogenesis of PCa include inflammation (Gueron et al.,
2012; Kazma et al.,
2012; Sfanos and de Marzo,
2012), oxidative stress and DNA damage (Miyake et al.,
2004; Lockett et al.,
2006; Battisti et al.,
2011; Gupta-Elera et al.,
2012), telomere shortening and telomerase activity (Kageyama et al.,
1997; Donaldson et al.,
1999; Fordyce et al.,
2005; Treat et al.,
2010; Xu et al.,
2011), genomic alterations (Boyd et al.,
2012; Nyquist and Dehm,
2013), and epigenetic modifications (Okino et al.,
2007; Goering et al.,
2012). Efforts have been made to achieve a deep understanding of these factors and consequently improve diagnosis and management of patients. However, the study of the trigger events for PCa initiation and development is compromised by the small number of PCa cell lines available for
in vitro studies, the majority of which are derived from metastatic and advanced cancers (Peehl,
2005).
Most tumors are androgen-dependent initially. However, as they develop they eventually become androgen-independent and progress to a hormone-refractory disease. This progression step is followed by metastasis formation (Heinlein and Chang,
2004).
PCa has an incredible propensity to metastasize, a consequence of several molecular mechanisms. Overall, these processes lead to local invasion, migration and site-specific establishment of metastases at secondary sites, usually in the bone, lung, or liver (Bubendorf et al.,
2000). Early steps of cancer progression include a down-regulation of cell-cell and cell-matrix characteristics. Malignant cells become motile and acquire the ability to destroy the extracellular matrix through degradative enzymes. In primary PCa, the expression of epithelial cadherin (E-cadherin), a transmembrane glycoprotein that is a key regulator of cell-cell binding and which is critical for epithelial to mesenchymal transition (EMT), was shown to be reduced. The down-regulation of E-cadherin enables EMT and consequently compromises cell-cell binding, so PCa can progress and metastasize. There is also evidence that abnormal signaling of β-catenin, a protein usually found complexed with cadherins, influences PCa progression due to dysfunction of the cadherin-catenin complex, an important early step in metastasis formation. Next, Ras and other GTP-binding proteins promote malignant cell motility and migration. As a solid tumor grows, malignant cells enter the circulation and rapidly bind to endothelial surfaces at secondary sites where they undergo transendothelial migration. In this process, several adhesive interactions occur involving selectin, integrin, platelet-endothelial cell adhesion molecule 1 (PECAM-1), vascular cell adhesion molecule 1 (VCAM-1), and other molecules. Once in a secondary site, tumor cells perturb the normal microenvironment and establish metastases (Clarke et al.,
2009).
2.3. Cell signaling pathways in prostate cancer
Cell signaling is essential for the normal function of the prostate gland. For instance, cell growth is controlled by insulin-like growth factor-1 (IGF-1) and fibroblast growth factor (FGF). Transforming growth factor-β (TGF-β) commands apoptosis, and vascular endothelial growth factor (VEGF) is particularly important for angiogenesis (Reynolds and Kyprianou,
2006). Several cell signaling pathways and crosstalks between them have also been implicated in PCa pathophysiology (Fig.
2). In this section, we briefly describe those that are most prominent.

Fig.2
Essential cell signaling pathways for prostate cancer (PCa) development and crosstalks between them
The interaction of circulating androgens (T/DHT) with AR is critical for PCa growth. Also, estrogens (E) and their corresponding receptors (ER) have a prominent role in prostate carcinogenesis, either by genomic or non-genomic actions. In advanced PCa, when the amount of available androgens is limited, other cytokines and growth factors promote synergic activities of AR. These molecules include TGF-β/BMPs, IL-6, EGF, IGF-1, VEGF, and FGF. Thus, androgen-responsive genes continue to be produced, although target genes differ between androgen-dependent and androgen-independent cells. In addition to their effects on the androgen pathway, these cell signals retain the ability to promote PCa overall survival, growth, and dissemination through alternative molecular targets. Grey arrows represent the androgen signaling in androgen-dependent PCa. Black arrows represent the prominent activated pathways in androgen-independent PCa. Dashed black arrows denote the crosslinks between cell signaling pathways. Black triangles symbolize up/down-regulation of molecules in PCa. Akt, protein kinase B; AR, androgen receptor; ARA55, androgen receptor associated protein 55; ARE, androgen response elements; BAG1L, Bcl-2-associated athanogene 1 long isoform; BMPs, bone morphogenetic proteins; BMPR, bone morphogenetic protein receptor; DDT, D-dopachrome tautomerase; DHT, dihydrotestosterone; D52, tumor protein D52; EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; FOXO1, forkhead box protein O1; GSTT2, glutathione S-transferase θ 2; HIF1A, hypoxia-inducible factor 1-α; HSP, heat shock proteins; IGF-1, insulin-like growth factor 1; IGFBP, IGF binding proteins; IGFR, insulin-like growth factor receptor; IL-6, interleukin 6; IL6R, interleukin 6 receptor; JAK, Janus kinase; KLK2, kallikrein-related peptidase 2; MAPK, mitogen-activated protein kinase; MEK, upstream kinases of mitogen-activated protein kinases; OAT, ornithine aminotransferase; PKC-δ, protein kinase C δ; P13K, phosphatidylinositol 3 kinase; PPPs, phosphoprotein phosphatases; PSA, prostate-specific antigen; PYCR1, pyrroline-5-carboxylate reductase 1; STAT3, signal transducer and activator of transcription 3; T, testosterone; TGF-β, transforming growth factor-β; TβR, transforming growth factor-β receptors; TMPRSS2, transmembrane protease serine 2; TRPV3, transient receptor potential cation channel subfamily V member 3; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor
2.3.1. Androgens
The androgen receptor (AR) signaling pathway is essential for the physiological development and maintenance of the prostate gland. In normal prostate cells, testosterone (T) or dihydrotestosterone (DHT), the more potent androgen, binds to AR, a nuclear receptor that in the absence of ligand is located in the cytoplasm. While no ligand binds to the AR, it remains in the cytoplasm complexed with heat shock proteins, cytoskeletal proteins, and chaperones that negatively regulate its nuclear translocation, transcriptional activity, and androgen dependence. Upon androgen binding, AR is translocated to the nucleus where it binds to androgen response elements in the promoter regions of target genes, regulating their expression (Fig.
2). Overall, it promotes cell proliferation, differentiation, apoptosis, and secretion (Nieto et al.,
2007; Lonergan and Tindall,
2011). AR activity is modulated by the action of co-receptors and by phosphorylation of both AR and AR co-receptors (Heinlein and Chang,
2004). Co-receptors can be either co-activators or co-repressors of AR activity. They include: proteins involved in the splicing process, RNA metabolism, DNA repair, endocytosis, and apoptosis; components of the chromatin remodeling complex, ubiquitin/proteasome pathway, and sumoylation pathway; histone modifier enzymes; chaperones and co-chaperones; proteins of the cytoskeleton; signal integrators and transducers; scaffolds and adaptors; cell cycle regulators; viral oncoproteins; nuclear receptor co-regulators; and kinases and phosphatases (Heemers and Tindall,
2007).
Most PCas remain responsive to androgen stimulation for their initial growth and so the blockade of the AR pathway might be sufficient to induce tumor regression (Yang et al.,
2005). In the early steps of malignant transformation, AR signaling changes from paracrine to autocrine and patients normally exhibit low levels of serum T and high expression levels of AR (Gao et al.,
2001; Vander Griend et al.,
2010). This could be caused by amplification or gain of function mutations in the AR genes, AR proteolytic processing to a constitutively active form, deregulation of AR co-factors, intratumoral androgen production, or crosslinks with other cell signals, such as growth factors, receptor tyrosine kinases, or protein kinase B (Akt) (Craft et al.,
1999; Buchanan et al.,
2001; Feldman and Feldman,
2001; Kang et al.,
2001; Meyer et al.,
2004). In contrast, a number of tumor cells present loss of AR expression. This phenomenon has been attributed to silencing by methylation, loss of the X chromosome, or a decrease in AR protein stability. Nevertheless, the real cause is still unclear (Heinlein and Chang,
2004). Notwithstanding, while PCa progresses, it eventually becomes refractory to androgens via diverse cell signaling pathway crosstalks that culminate in abnormal AR signaling (Nieto et al.,
2007).
The mechanisms underlying the development of androgen-independence have been extensively investigated. Splice variants of AR were identified and shown to be important for the development of androgen-independence. The characterizations of AR
3 and AR
8 in hormone insensitive PCa cells have been of particularly interest (Guo et al.,
2009; Yang et al.,
2011). AR
3 is constitutively active and is an androgen-independent transcription factor. It was shown to be expressed at higher levels in androgen-independent cells compared to androgen-sensitive cells, resulting in the promotion of cancer cell proliferation (Guo et al.,
2009). AR
8 is a less abundant AR splice variant that localizes in the plasma membrane and acts mainly through non-genomic mechanisms, by associating with epidermal growth factor receptor (EGFR). It also enhances Scr activation, AR tyrosine phosphorylation, and the association between AR and EGFR. Overall, AR
8 potentiates AR-mediated proliferative and survival responses to hormones and growth factors (Yang et al.,
2011). Moreover, it was recently shown that truncated forms of AR with intact NH
2-terminal and DNA binding domains are constitutively active and able to maintain androgen-independent transcriptional activation of endogenous AR target genes, thus supporting androgen-independent growth of PCa cells (Chan et al.,
2012). The expression of these forms is associated with a poor prognosis and they have been recognized as potential therapeutical targets to treat androgen-independent PCa (Hörnberg et al.,
2011).
Modern techniques, such as chromatin immunoprecipitation display and microarrays, have been applied to unveil the AR target genes involved in transcriptional regulation in PCa. Genes are expressed differently in normal prostate and PCa tissues. Transmembrane protease serine 2 (TMPRSS2) and its fusions with E-twenty six (ETS) transcription factor family members, mainly v-ets erythroblastosis virus E26 oncogene homolog (ERG) or its variant 1 (ETV1), kallikrein-related peptidase 2 (KLK2), and tumor protein D52, are among the genes expressed by androgen-sensitive cells when stimulated by androgens (Lonergan and Tindall,
2011; Marques et al.,
2011). Genes identified in androgen-independent PCa cells include
D-dopachrome tautomerase, protein kinase C δ, glutathione
S-transferase θ 2, transient receptor potential cation channel subfamily V member 3, pyrroline-5-carboxylate reductase 1, and ornithine aminotransferase (Jariwala et al.,
2007). Despite the advances in this field, much remains to be clarified.
2.3.2. Estrogens
Circulating levels of estrogens in males tend to increase during aging. Besides, testosterone can also be continuously converted to 17β-estradiol by aromatase within the prostate stroma. With aging, there are decomposition of androgens and a predominance of estrogens, which can regulate prostate growth either directly or indirectly via regulation of other hormones, such as prolactin and luteinizing hormone (Huggins and Hodges,
1972; Lee et al.,
1981; Marino et al.,
2006; Carruba,
2007). Thus, the estrogen receptor (ER) signaling pathway has been receiving increasing attention as an intervenient in PCa carcinogenesis (Carruba,
2007).
ERs belong to a family of nuclear transcription factors which are ubiquitously expressed. Different genes encode two ER subtypes, ERα and ERβ, which exhibit several isoforms (Kuiper et al.,
1996; Fixemer et al.,
2003). In the absence of ligand, ER is found in the nucleus complexed with inhibitory proteins. Upon ligand binding, ER is activated and binds to estrogen response elements in the promoter region of target genes (Fig.
2). In addition to this “genomic action”, ER also exhibits a “non-genomic action” via activation of other cell signaling pathways: phospholipase C/protein kinase C, Ras/Raf/mitogen-activated protein kinase (MAPK), phosphatidyl inositol 3 kinase (PI3K)/Akt, and cyclic-adenosine monophosphate (cAMP)/protein kinase A (PKA) (Marino et al.,
2006; Carruba,
2007). There is also evidence that ER activation can occur in a ligand-independent manner via MAPK and cAMP/PKA (Coleman and Smith,
2001).
Estrogens and associated signaling pathways appear to play a critical role in PCa development and progression. Metabolic activation of estrogens to genotoxic metabolites, such as 2- and 4-hydroxyl catechol estrogens, induces DNA damage, oxidative stress, and the formation of high levels of reactive oxygen species (Yager,
2000; Cavalieri and Rogan,
2006; Singh et al.,
2008). Concerning the ERs, ERβ is progressively lost during human prostate carcinogenesis. ERβ isoforms influence the human prostate in different ways depending on the spliced variants expressed and the use of alternative promoters (Horvath et al.,
2001; Fixemer et al.,
2003; Leung et al.,
2010b). For instance, ERβ1 inhibits the proliferation of prostate epithelial cells and EMT, whereas ERβ2/5 promotes metastasis formation (Leung et al.,
2010a; Mak et al.,
2010; McPherson et al.,
2010). In contrast, there is a gradual increase in the expression of ERα from PIN to metastatic lesions. Also, evidence suggests that during PIN, ERα is expressed in both stromal and luminal cells (Ricke et al.,
2008). In combination with AR, this receptor stimulates proliferation and differentiation of PCa cells (Bonkhoff et al.,
1999). Also, it was found that the enzyme that catalyzes estradiol production from testosterone, aromatase (CYP19), is altered in PCa tissues (Ellem et al.,
2004).
2.3.3. TGF-β
TGF-β belongs to the TGF-β superfamily of cytokines that also includes activins and bone morphogenetic proteins (BMPs). Members of this protein family are involved in the regulation of several physiological processes, such as cell proliferation, differentiation, apoptosis, adhesion, chemotaxis, organogenesis, and angiogenesis (Korrodi-Gregorio et al.,
2012).
In this signaling pathway, ligands bind to the TGF-β receptor II (TβRII), a transmembrane receptor, which in turns recruits the TGF-β receptor I (TβRI) to form a heteromeric complex. TβRIs are also referred to as activin receptor-like kinases (ALKs) and they show different affinities for ligands (Derynck and Feng,
1997). TβRII phosphorylates and activates TβRI and the intracellular signaling proceeds due to phosphorylation of Smads. Smads are classified into three groups: (1) receptor-associated Smads (R-Smads)—Smad-1, -2, -3, -5, -8; (2) common Smad (Co-Smad)—Smad-4; and (3) inhibitory Smads (I-Smad)—Smad-6, -7. TβRI phosphorylates and activates R-Smads which bind with high affinity to Co-Smad. The heteromeric complex formed translocates into the nucleus where it binds to transcription promoters or co-factors, enabling DNA transcription. I-Smads provide a mechanism for negative feedback by preventing the interaction between R-Smads and TβRI or Co-Smad (Bello-DeOcampo and Tindall,
2003; Korrodi-Gregorio et al.,
2012).
Components of the TGF-β signaling pathway have been implicated in PCa regulation, with either tumor suppressor or tumor promoter activities being attributed to it. More specifically, TGF-β signaling exhibits growth inhibitory effects in the early stages of PCa and promotes malignancy in later stages. Disruption of TGF-β signaling is referred to as a metastasis promoter (Tu et al.,
2003). The loss or reduction of sensitivity to the TGF-β inhibitory effect and the acquired ability to express higher levels of TGF-β are in part explained by the ability of cells to overwhelm the down-regulatory effects of androgens. These phenomena appear to be associated with the transformation of a prostate tumor from benign to malignant (Saez et al.,
1998; Lee et al.,
1999).
The adult prostate gland expresses mostly TGF-β1 when compared to TGF-β2 and TGF-β3 isoforms (Bello-DeOcampo and Tindall,
2003). TGF-β1 acts as a tumor suppressor in normal prostate epithelial cells. During progression to metastatic PCa, TGF-β1 overexpression, due to androgen withdrawal, enhances oncogenesis (Merz et al.,
1994; Zhu and Kyprianou,
2008). In contrast, AR expression directly down-regulates or inhibits the transcriptional activity of TGF-β1 signaling and reduces its growth effects (when DHT is absent) (Zhu and Kyprianou,
2008). PCa cells are able to produce the bioactive form of TGF-β1 by themselves. Instead of the growth inhibitory effect that TGF-β1 exhibits in the normal prostate gland, in PCa cells it enhances prostate tumor growth, angiogenesis, immunosuppression, remodeling of the extracellular matrix, and metastasis formation (Barrack,
1997; Festuccia et al.,
1999). However, recent data also show that TGF-β1 decreases cell viability and induces apoptosis in invasive tumor cells via activation of p38 MAPK and c-Jun N-terminal kinases (JNK), which in turn promote the expressions of the cleaved caspases-8, -9, and -3 (Al-Azayzih et al.,
2012). The roles of TGF-β2 and -β3 remain unclear.
Regarding the TGF-β receptors, there is a decrease in both TβRI and TβRII receptors with PCa progression (Guo et al.,
1997; Kim et al.,
1998). This decrease implicates a reduction in the TGF-β growth inhibitory response in tumor cells. Endoglin, an auxiliary receptor of the TGF-β signaling whose expression is lost during PCa progression, was shown to suppress PCa cell motility through activation of the ALK2/Smad-1 pathway (Craft et al.,
2007).
BMPs are bone inductive factors of the TGF-β superfamily that have been implicated in bone metastasis formation (Ye et al.,
2007a). BMP-6 expression is absent in patients with benign prostatic hyperplasia (BPH), but its expression has been reported in malignant prostatic epithelial cells, mainly in primary PCa with established secondary skeletal metastases (Hamdy et al.,
1997). High levels of BMP-6 signaling, a consequence of its increased expression and the decreased expression of its inhibitors, might promote the development of PCa metastases (Yuen et al.,
2008). This growth factor may have a direct inductive role in PCa-associated bone metastases (Dai et al.,
2005). The increased metastatic potential promoted by BMP-6 is at least in part because of the increase in metaloproteinases and Id-1 (Darby et al.,
2008), a downstream BMP target that was previously shown to be indicative of a worse prognosis in early-stage cervical cancer and epithelial ovarian tumors (Schindl et al.,
2001;
2003). In contrast, BMP-9 and BMP-10 may function as tumor suppressors and apoptosis regulators in PCa. Their expression is decreased or even absent in PCa. It was demonstrated that their overexpression in PCa cells prevented
in vitro growth, cell-matrix adhesion, invasion, and migration of cells (Ye et al.,
2008;
2009). BMP-9 up-regulates and activates prostate apoptosis response-4 through a Smad-dependent pathway, thus promoting apoptosis of PCa cells (Ye et al.,
2008). BMP-10 acts, at least in part, through the Smad-independent pathway X-chromosome-linked inhibitor of apoptosis protein/transforming growth factor-β-activated kinase-1/extracellular regulated kinase (XIAP-TAK1-ERK) (Ye et al.,
2009). Another BMP predominantly expressed in normal prostate tissue is BMP-7, whose expression is androgen-dependent (Masuda et al.,
2004). It is important for controlling noggin and follistatin expression, antagonists of the BMP signaling pathway, and therefore the regulation of the invasion, motility, and adhesion of PCa cells (Ye et al.,
2007b). The loss of endogenous BMP-7 during PCa progression is associated with increased invasiveness and motility (Ye et al.,
2007b). The expression of BMP-2 also decreases with the progression to malignancy and the decrease correlates with an increasing Gleason score (Horvath et al.,
2004). Its function is dependent on androgens: in the presence of androgens it inhibits the growth of PCa cells, while in the absence of androgens it stimulates cell growth (Langenfeld and Langenfeld,
2004).
Nodal, another member of the TGF-β superfamily, is also expressed in some PCa cell lines. It inhibits proliferation and induces migration depending on the stage in cancer progression and the cell microenvironment (Vo and Khan,
2011).
Several studies also revealed an up-regulation in the expressions of Smad-2, -3, -4, -6, and -7 after androgen deprivation, while the expressions of Smad-1, -5, and -8 are down-regulated in advanced PCa (Romero et al.,
2010; Lakshman et al.,
2011). The activity of Smad intracellular signaling in PCa has been explored at length. Smad-7 is an effector in TGF-β1-induced apoptosis and its overexpression in PCa cells induces apoptosis (Brodin et al.,
1999; Landstrom et al.,
2000). More specifically, Smad-7 acts as a scaffolding protein and facilitates the activation of the p38 MAPK pathway mediated by TGF-β-activated kinase 1 and MAPK-3 (Edlund et al.,
2003). Then, p38 modulates the increased adhesion of metastatic PCa cells through phosphorylation and activation of Smad-3 (Hayes et al.,
2003). It was also shown that Smad-7 interacts with growth arrest and DNA damage protein (GADD34), a regulatory/targeting subunit of phospho-protein phosphatase 1 (PPP1). The catalytic subunit of PPP1 is then recruited to the TβRI-Smad7-GADD34 complex and dephosphorylates TβRI, establishing a negative feedback of the TGF-β pathway (Shi et al.,
2004). Overexpression of Smad-7 may also be the reason for the decreased levels of phosphorylated Smad-2 observed in PCa. However, this decrease implicates a reduction in the nuclear Smad-4. Smad-4 (either dependent on or independent of Smad-3) interacts with the DNA-binding and the ligand-binding domains of AR, and apparently modulates DHT-induced AR transactivation (Zhu and Kyprianou,
2008). Loss of nuclear Smad-8 during PCa progression has also been reported (Horvath et al.,
2004; Perttu et al.,
2006).
2.3.4. IGF-1 and PI3K/Akt
IGF-1 is produced by prostatic stromal cells in response to androgen stimulation. This constitutes a paracrine signaling, stimulating the surrounding prostatic epithelial cells that increase cell proliferation. In normal cells, this pathway is repressed by IGF binding proteins (IGFBPs), which bind with high affinity to IGF-1 and prevent its interaction with the receptor. Receptor substrates include PI3K and Ras-MAPK pathways (Pollak,
2008).
The sustained activation of the IGF-1 signaling pathway is responsible for the proliferation of PCa cells. Moreover, a direct correlation was observed between high plasma IGF-1 levels and PCa progression (Stattin et al.,
2004). The IGF-1 receptor is overexpressed in primary PCa, in part because of the stimulation by early growth response-1 (EGR-1) (Ma et al.,
2012). In general, the effects of this pathway in PCa are due to amplification of the Akt gene or its upstream kinases, such as PI3K, or deletion/mutation of negative regulators, such as PTEN, PPP1, and phospho-protein phosphatase 2 (PPP2) (Li et al.,
2005).
Upon phosphorylation, PI3K becomes active and promotes cell proliferation and survival by regulating downstream targets. One most prominent target is Akt that belongs to the serine/threonine protein kinase B family. PI3K mediates both G1 cell cycle progression and cyclin expression by activating the Akt/mammalian target of the rapamycin (mTOR)/p70 (S6K) signaling pathway in PCa cells (Gao et al.,
2003). When phosphorylated, the activated Akt (pAkt) inhibits the glycogen synthase kinase 3β (GSK3β) which normally increases the degradation of cyclin D1, and consequently prevents the up-regulation of cell proliferation. pAkt also induces the production of desmoplakin, vimetin, and metaloproteinases and thus regulates EMT and cell invasion (Bellacosa and Larue,
2010). A recent study revealed that the expression of E-cadherin is controlled by the human metastasis-associated gene 1 (MTA1) through a pAkt-dependent mechanism (Wang et al.,
2012). Another target of PI3K is the proapoptotic protein Bad, which is inhibited upon phosphorylation, thereby blocking apoptosis of PCa cells. The Ras/MAPK pathway has a similar effect on the bad protein, leading to cell survival and proliferation (Moschos and Mantzoros,
2002).
2.3.5. Other cell signalings
Epidermal growth factor (EGF), its receptor (EGFR), and the subsequent intervenients of this pathway promote cell growth, proliferation, and differentiation. Frequently, EGF and EGFR are up-regulated in advanced PCa. Activation of this signaling in PCa cells induces activation of the MAPK pathway that in turn down-regulates AR. Also, AR activates MAPK and EGFR. Moreover, EGF is able to induce up-regulation of interleukin-6 (IL-6) (Morgan et al.,
2009).
The FGF protein family is involved in growth, proliferation, development, and angiogenesis. Altered expression in FGFs and respective receptors correlates with PCa progression towards an androgen-independent state. FGF-2 (or basic FGF, bFGF), FGF-7, and FGF-8 are the growth factors with a more established role in PCa. All three were shown to be overexpressed in hyperplasic tissues.
FGF-2 is a mitogenic agent for prostatic stromal cells, acting mainly in an autocrine manner. It contributes to cell differentiation, migration, and angiogenesis (Reynolds and Kyprianou,
2006). FGF-2 expression and release were shown to up-regulate TβRII/Smad3, enabling the angiogenic and tumor promoting effect observed in reactive stroma (Yang et al.,
2008). In contrast, FGF-7 acts as a mitogen for prostatic epithelial cells, exerting its effect in a paracrine manner (Reynolds and Kyprianou,
2006). The function of FGF-8 remains to be elucidated. However, its expression in PCa is at least in part regulated by AR (Gnanapragasam et al.,
2002). Recent studies have implicated other members of the FGF superfamily in PCa pathogenesis. A case-control study reported that the Gly-388Arg polymorphism of the FGF receptor-4 (FGFR4) was a contributing factor to PCa susceptibility (Liwei et al.,
2011). Combined with the expression of proteins that stabilize FGFR4, this polymorphism endorses a more stable signal, thus promoting PCa progression (Wang et al.,
2008). FGF-9 stimulates proliferation and invasion in PCa cells (Teishima et al.,
2012). FGF-19 is expressed in primary and metastatic PCa tissues where it acts as an autocrine growth factor (Feng et al.,
2013).
VEGF is a cytokine involved in cellular proliferation, migration, and angiogenesis. VEGF levels are higher in PCa compared to normal or benign hyperplasia (Ferrer et al.,
1997) and represent the strongest stimulus for angiogenesis in endothelial PCa cells (Trojan et al.,
2004). Moreover, the expressions of VEGF ligands and receptors (VEGFRs) are higher in bone metastasis sites than in the primary tumor. Through VEGF receptor-2 (VEGFR-2), VEGF regulates the migratory responses of PCa cells to fibronectin and bone sialoprotein, proteins of the ECM (Chen et al.,
2004). The type of VEGFR expressed in PCa cells influences cell growth, invasion, and metastasis formation. For instance, Qi et al. (
2003) found that cell lines expressing NP-1 receptor had lower levels of migration, with and without VEGF, compared to those that did not express NP-1. On the other hand, the increased expression levels of VEGF isoform C and its receptor VEGFR-3 are associated with lymph node metastasis formation. This isoform also increases the expression of Bcl-2-associated athanogene 1 long isoform (BAG-1L), an AR co-activator that facilitates AR transactivation (Jennbacken et al.,
2005; Chetram et al.,
2011).
Interleukins, especially IL-6, have been shown to play an important role in PCa development and progression. Levels of IL-6 are increased in PCa and correlate with worse progression scenarios (Azevedo et al.,
2011). IL-6 binds to its receptor and activates Janus kinase (JAK) cell signaling, but also MAPK and PI3K pathways, depending on the cell type. Subsequent to JAK activation, the signal transducer and activator of transcription 3 (STAT3) is phosphorylated and translocated into the nucleus. STAT3 phosphorylates the AR at serine-772, a process essential for its transcriptional activity (Aaronson et al.,
2007). Increasing evidence suggests a role for STAT3 in the metastatic behavior of PCa cells (Abdulghani et al.,
2008; Gu et al.,
2010).
2.3.6. Phosphatases: essential regulators of cell signaling
Many cellular processes are controlled by phosphorylation/dephosphorylation of structural or regulatory proteins. The reversible phosphorylation system encompasses kinases that add a negatively charged phosphate group mainly to serine, threonine or tyrosine residues, and phosphatases that remove those phosphate groups (Fardilha et al.,
2011). In cancer, imbalances in protein phosphorylation systems appear to be an important pathophysiologic mechanism. Since the activation of several kinases results in stimulation of cell signaling pathways that potentiate cell growth and proliferation, it is not surprising that tumor suppressive functions have been attributed to phosphatases (Fardilha et al.,
2010).
In PCa the tumor suppressor gene that encodes PTEN is often lost. PTEN is a lipid phosphatase that negatively regulates PI3K/Akt signaling by dephosphorylation of phosphatidylinositol 3,4,5-phosphate (PIP3). Thus, mutations of PTEN are frequently associated with cancer development (Salmena et al.,
2008). Another element that directly suppresses PTEN expression in PCa cells is the microRNA miR-153 (Wu et al.,
2012). Abnormalities in PTEN are observed not only in PCa but also in several cases of PIN, suggesting that PTEN and Akt might be critical for early PCa development (Squire,
2009). PTEN loss enhances the activation of several signaling pathways which are normally deregulated during PCa, including PI3K/Akt and MAPK pathways (Goc et al.,
2011). The PTEN/PI3K/Akt pathway was shown to be essential for the maintenance of PCa stem-like cells, being important to their survival and proliferation (Dubrovska et al.,
2009). In the absence of PTEN, cells also present high levels of mTOR activity which results in uncontrolled growth (Majumder and Sellers,
2005). Moreover, loss of PTEN seems to mediate bone tropism in PCa metastases, at least through Rac1, a small GTPase that is a PTEN effector (Wu et al.,
2007). Overall, patients with PCa that exhibit a PTEN mutation have a higher Gleason score, a poorer prognosis, and a greater rate of metastases (Pourmand et al.,
2007). Together with VEGF receptor-2, PTEN regulates PCa proliferation and cell adhesion to fibronectin (Chen et al.,
2004).
In addition to PTEN, PPP1 and PPP2 modulate PCa progression by interacting with key proteins. In PCa, both phosphatases seem to act as tumor suppressors. PPP1 and PPP2 activities were previously shown to be needed to maintain endothelial cells in a resting state (Gabel et al.,
1999). Inhibition of both phosphatases by caveolin-1 led to increased activities of PDK1, Akt, and ERK1/2. This, in turn, promoted tumor cell survival (Li et al.,
2003). Recently, the activation of phospho-protein phosphatase 5 (PPP5) by caveolin-1 was also confirmed (Taira and Higashimoto,
2013). The complex formed between PPP1 and its nuclear inhibitor was recently described as a regulator of PCa direct cell migration via up-regulation of Cdc42 (Martin-Granados et al.,
2012). PPP1 also interacts with AR, regulating its stability and nuclear localization through dephosphorylation at Ser650. PPP1 inhibition enhances proteasome-mediated AR degradation, while PPP1 overexpression increases AR expression and markedly enhances AR transcriptional activity in PCa cells (Chen et al.,
2009). The activation of PPP1 in malignant cells is potentiated by the down-regulation of Fer, a tyrosine kinase. PPP1 also affects TGF-β signaling in PCa cells through the complex formed with GADD34 and Smad-7, as mentioned above. PPP1 dephosphorylates and activates phosphorylated retinoblastoma protein (Rb), leading to its growth suppressive state and cell-cycle arrest in malignant cells (Pasder et al.,
2006). The expression of the catalytic subunit of PPP2 and its activity were shown to be down-regulated in androgen-independent PCa cells compared to androgen-dependent cells. This is particularly interesting as PPP2 is able to sustain the growth of androgen-dependent cells under androgen-deprivation by relieving the androgen deprivation-induced cell-cycle arrest and preventing apoptosis (Bhardwaj et al.,
2011).
3. Conventional diagnosis and monitoring of prostate cancer
Currently, the screening for PCa essentially involves digital rectal examination (DRE), determination of serum concentrations of PSA, and transrectal ultrasonography (TRUS). Table
1 describes the main advantages and disadvantages of these techniques.
Table 1
Presentation of the three principal methods used for the current screening and diagnosis of prostate cancer: digital rectal examination (DRE), prostate-specific antigen (PSA) measurements, and transrectal ultrasonography (TRUS)
| Method |
Advantage |
Disadvantage |
Reference |
| DRE (physical exam) |
Safe exam; May detect PCa in men with normal PSA levels and small tumors; Relatively inexpensive |
Most palpable cancers are not early cancers; Cancers can be located in distant regions of the gland, and so are not palpable; Conclusion from the exam is highly dependent on the interpretation of the examiner; Its sensitivity and specificity are low and dependent on tumor stage |
Okotie et al., 2007
|
| PSA measurement (biochemical test) |
Safe exam; Used for screening, early detection, and prognosis; Good sensitivity; Relatively inexpensive |
Low specificity, thus leading to high number of false positives; Detection rate of PCa when using PSA levels combined with DRE depends on both the PSA threshold used on biopsy and the patient’s age; Correlation between grade and progression has been referred to as feeble in some ranges of detection |
Stamey et al., 2002; Bickers and Aukim-Hastie, 2009; Shteynshlyuger and Andriole, 2010
|
| TRUS (imagiologic exam) |
Enables early diagnosis; Useful to guide biopsies; High sensitivity |
Invasive procedure; Poor specificity; Time-consuming technique |
Shteynshlyuger and Andriole, 2010
|
Definitive diagnosis of PCa always requires the presence of adenocarcinoma in prostate biopsies or operative specimens. Prostate biopsy is an essential procedure for PCa diagnosis, presenting 100% specificity and virtually no false positives. Although major complications have not been associated with this procedure, it is an invasive technique and so its clinical application in daily screening is conditioned (Raaijmakers et al.,
2002). Moreover, the number of false negative cases is still higher than desirable (Haas et al.,
2007). Histopathological examination of prostate specimen enables both grading and determination of tumor extent. The Gleason score is the standard system used by clinicians to rate PCa. This system classifies the biopsy specimen from 1 to 5, with 5 being the most aggressive with no glands being recognized. The final Gleason score is a sum of the most common primary and secondary grades. Sometimes a tertiary grade can also be given (Borley and Feneley,
2008).
4. Emerging biomarkers: new tools to improve prostate cancer diagnosis and management
A biomarker, as defined by the National Cancer Institute, is “a biological molecule found in blood, other body fluids, or tissues that is a sign of a normal or abnormal process or of a condition or disease” (
http://www.cancer.gov/dictionary?cdrid=45618). These molecules are useful tools in supporting several steps of a patient’s management: prevention, screening, diagnosis, prognosis, prediction of treatment effectiveness, and monitoring responses to treatment. The establishment of a specific panel of biomarkers, in either tissue or body fluids, might complement the routinely applied diagnostic techniques to achieve earlier and more accurate diagnoses. This is a major challenge as PCa is a silencing disease in the early stages, hence not presenting symptoms until it becomes locally advanced or metastasizes (Smith et al.,
2003). In addition, to enable the evaluation of the course of the disease, prognostic biomarkers might support treatment decisions. Predictive biomarkers are helpful in choosing the treatment type. Biomarkers can also aid drug development, and the advantage of their use in clinical trials has been widely explored (Rolan,
1997; Alaoui-Jamali and Xu,
2006; Committee on Developing Biomarker-Based Tools for Cancer Screening, Diagnosis, and Treatment,
2007).
Nowadays, only two biomarkers are approved by the FDA for use in PCa management: total PSA (tPSA) and free PSA (fPSA) (Rhea and Molinaro,
2011). Increased levels of serum tPSA are associated with PCa. tPSA has been widely used for screening, diagnosis, and monitoring of the disease, especially for the detection of recurrent disease after treatment (Makarov et al.,
2009). However, as previously noted, it lacks specificity as conditions such as prostate infection, irritation, benign prostatic hypertrophy, recent ejaculation, or medical interventions can also cause an upsurge in tPSA levels (Makarov et al.,
2009; Rhea and Molinaro,
2011). fPSA corresponds to the portion of secreted PSA that does not form complexes with serum antiproteases. Patients with PCa present a lower proportion of fPSA to tPSA (%fPSA), and so this percentage can be used to reduce unnecessary biopsies and to follow the progression of the disease. The %fPSA is, nevertheless, modulated by prostate volume, patient age, tPSA levels, and prostatic manipulation (Makarov et al.,
2009).
Prior to PSA, prostatic acid phosphatase (PAP) was extensively used as a serum prognostic biomarker for PCa. Higher levels of expression and activity of PAP are associated with PCa with established metastases on bone. Also, post-treatment PAP levels are predictive of the clinical outcome.
Efforts have been made to discover new tissue biomarkers and molecules measurable in body fluids with high sensitivity and specificity. Table
2 summarizes some of the most relevant molecular markers that can be used in the context of PCa.
Table 2
Emerging biomarkers for diagnosis, prognosis, and prediction of prostate cancer (PCa): a brief characterization of each biomarker and observations that justify its use in the management of patients with PCa
| Biomarker |
Characterization |
Observation in PCa |
Detection |
Use |
Reference |
| α methylacyl coenzyme A racemase (AMACR) |
Protein involved in the β-oxidation of branched fatty acids |
Overexpressed in PCa specimens, with levels greater than PSA. mRNA levels of AMACR and survivin together aid the identification and risk stratification of patients with PCa |
Tissue |
Diagnosis, predictive |
Rubin et al., 2002; Liu et al., 2007; Al-Maghrebi et al., 2012; Yap et al., 2012
|
| Adenomatous polyposis coli (APC) |
Tumor suppressor gene |
Loss of heterozygosity, lower expression, and methylation of APC are described in PCa |
Tissue, serum, urine |
Diagnosis |
Gao et al., 1995; Liu et al., 2011; Chen Y. et al., 2013
|
| Bone turnover markers (BTMs) |
Markers of bone formation, such as osteocalcin, total alkaline phosphatase activity, and serum bone-specific alkaline phosphatase, as well as, bone resorption, including C-telopeptides of type I collagen |
Higher levels of BTM are associated with negative clinical outcomes in patients presenting bone metastases. A reduction in their levels after treatment is related to patient survival, both in androgen-dependent or -independent PCa |
Serum |
Diagnosis, prognosis, predictive |
Garnero et al., 2000; Garnero, 2001; Hegele et al., 2007; Som et al., 2012
|
| Circulating tumor cells (CTCs) |
Tumor cells that enter the bloodstream as a consequence of tumor growth. Responsible for establishing metastases |
Higher amounts of CTCs are related to worse prognoses. They also provide information about other molecular markers, including TMPRSS2-ERG and EMT-related genes. Commercial kits are already available to count CTCs in metastatic PCa |
Blood |
Prognosis |
Danila et al., 2011; Chen C.L. et al., 2013; Thalgott et al., 2013
|
| Endoglin (CD105) |
Transmembrane glycoprotein that constitutes a co-receptor for TGF-β signaling |
Primarily detected in newly formed vessels, being related to the angiogenic process. Preoperative serum levels of endoglin correlate with pelvic lymph node metastasis after radical prostatectomy |
Tissue, serum |
Prognosis, predictive |
Wikstrom et al., 2002; Karam et al., 2008
|
| Early prostate cancer antigen (EPCA) |
PCa-specific nuclear matrix protease |
PCa patients have higher serum levels of EPCA. The highest preoperative serum levels correspond to metastatic PCa cases |
Serum |
Diagnosis, prognosis |
Zhao et al., 2010; 2012
|
| Golgi membrane protein 1 (GOLM1) |
Type II Golgi transmembrane protein that collaborates in protein synthesis and transport of protein cargo |
Up-regulation of GOLM1 expression in PCa compared with normal prostate and BPH tissues |
Tissue, urine |
Diagnosis |
Varambally et al., 2008; Li et al., 2012
|
| Glutathione S-transferase-P1 gene (GSTP1) |
Encodes a detoxifying enzyme that protects DNA from free radical damage |
Serum methylation profiles differ between healthy subjects, BPH, and PCa patients. PIN and PCa tissues present loss of GSTP1 expression due mainly to promoter hypermethylation. Together with APC levels it might improve the distinction between PCa and BPH. Methylation status of GSTP1 gene and Gleason score are strictly related |
Tissue, serum |
Diagnosis, prognosis |
Bernardini et al., 2004; Liu et al., 2007; Bryzgunova et al., 2008; Karam et al., 2008; Woodson et al., 2008; Yoon et al., 2012
|
| Hepsin |
Type II transmembrane serine protease |
Overexpressed in PCa when compared to healthy or BHP tissues |
Tissue |
Diagnosis |
Klezovitch et al., 2004; Kelly et al., 2008; Goel et al., 2011; Pace et al., 2012
|
| Human kallikrein 2 (KLK2) |
Serine protease that belongs to the human kallikrein family, as well as PSA. It is predominantly expressed in the prostate under androgen control |
Overexpressed in HGPIN and PCa when compared to healthy tissue. Increased serum levels are associated with worse outcomes |
Tissue, serum |
Diagnosis, prognosis |
Darson et al., 1997; Raaijmakers et al., 2007; He et al., 2009
|
| Ki-67 |
Mitotic index that regulates the cell-cycle |
Increased Ki-67 staining index correlates with higher Gleason score. Together with other molecular markers, it helps to determine the risk of PCa progression |
Tissue |
Prognosis |
Steuber et al., 2007; Madani et al., 2011; Josefsson et al., 2012; Fisher et al., 2013
|
| Lactate dehydro-genase (LDH) |
Metabolic enzyme that participates in glycolysis and gluconeogenesis pathway |
Increased levels of LDH are associated with poor clinical outcomes in PCa patients and aggressive phenotypes in patients with bone metastases. Expression of the gene encoding LDH H subunit is lost through promoter methylation |
Blood |
Prognosis |
Leiblich et al., 2006; Naruse et al., 2007
|
| MicroRNAs |
Noncoding regulatory RNAs, such as miR-21, miR-75, miR-141, and miR-221 crucial in development and post-transcriptional processes |
Increased levels in patients with metastatic PCa. Higher levels of miR-75 inhibit Sec23A protein, promoting cell proliferation |
Tissue, serum |
Diagnosis, prognosis |
Mattie et al., 2006; Mitchell et al., 2008; Szczyrba et al., 2011; Yaman Agaoglu et al., 2011
|
| p63 |
Gene that belongs to the p53 gene family. It inhibits PCa metastases through regulation of miR-205 expression |
The majority of PCa tissues do not express p63, unlike normal and preneoplastic tissue. Aberrant expression of p63 is associated with oncogenic progression and survival |
Tissue |
Diagnosis |
Parsons et al., 2001
|
| Prostate cancer antigen 3 (PCA3) |
PCa-specific gene that modulates the transcriptional activity of androgen receptor target genes. Involved in PCa survival |
Highly overexpressed in PCa tissues, but not in normal or BPH tissues. In contrast to PSA, it is a marker independent of prostate size. As observed in PCa tissues, PCA3 is up-regulated in patients’ urine. Combining the PCA3 level with the PSA level may decrease the number of unnecessary prostate biopsies |
Tissue, urine |
Diagnosis |
Bussemakers et al., 1999; Crawford et al., 2012; Ferreira et al., 2012; Yutkin et al., 2012
|
| Phospho-choline-containing phospholipid (PcCP) |
Lipids are involved in several functions, including cellular energy storage, structure, and signaling |
Patients with PCa have increased plasma lipid concentrations of almost all classes of lipids. Levels of lipids, such as myristic acid, α-linolenic acid, eicosapentaenoic acids, and cholesterol modulate the risk of PCa |
Serum |
Diagnosis |
Zhou et al., 2012
|
| Prostate-specific antigen (PSA)-derived markers |
Include PSA density, PSA velocity, pro-PSA, and PSA doubling time |
Patients with PCa present increased PSA density, higher PSA velocity, lower PSA doubling time, and higher levels of pro-PSA |
Serum |
Predictive, prognosis |
Makarov et al., 2009
|
| Phosphatase and tensin homolog (PTEN) |
Tumor suppressor gene that encodes the phosphatase responsible for the conversion of PIP3 to inactive phosphatidylinositol (4,5) biphosphate |
PTEN deletion is common in PCa and is associated with decreased expression of PTEN and AR, which represents a poorer outcome |
Tissue |
Prognosis |
Lotan et al., 2011; Antonarakis et al., 2012; Choucair et al., 2012
|
| Transforming growth factor-β1 (TGF-β1) |
Regulator of cellular proliferation, differentiation, and angiogenesis |
Decreased expression levels in PCa. Polymorphisms also aid the determination of PCa progression (single nucleotide polymorphisms at C-509T correlate with lower risk of aggressive PCa) |
Tissue |
Predictive |
Soulitzis et al., 2006; Faria et al., 2007; Brand et al., 2008
|
| Trans-membrane protease serine 2-v-ets erythroblastosis virus E26 oncogene homolog (avian) (TMPRSS2-ETS gene fusion) |
Fusion of TMPRSS2 gene, encoding a serine protease, with an ETS family member oncogene, such as ERG, culminates in ETS oncogene being overexpressed under androgen control |
TMPRSS2-ERG fusion is the most frequent. TMPRSS2-ETS gene fusion is common in PCa tissues and precancerous lesions closed to PCa. TMPRSS2-ETS gene fusion together with PCA3 levels supports risk stratification |
Tissue, urine |
Predictive, diagnosis, prognosis |
Tomlins et al., 2005
|
| Urokinase plasminogen activator (uPA) and urokinase plasminogen activator receptor (uPAR) |
Cell signaling involved in the degradation of the extracellular matrix and basement membrane |
Increased expression of uPA is associated with bone metastases. High serum levels of the uPAR forms correlate with poorer outcomes |
Tissue, serum |
Prognosis, predictive |
Ohta et al., 2003; Kumano et al., 2009; Almasi et al., 2011
|
5. Current options and challenges in prostate cancer treatment
PCa treatment must be adjusted according to: (1) the tumor stage and grade, (2) PSA levels, and (3) the estimated baseline for patient life expectancy (Heidenreich et al.,
2012). In terms of response to treatment, four stages can be identified while the tumor progresses: hormone-sensitive, androgen-independent, symptomatic metastatic, and advanced metastatic or anaplastic. Nowadays, treatment options are available for each stage. However, the ideal therapy has not yet been found and treatment effectiveness varies with cancer progression (Oudard,
2013).
Localized tumors can be efficiently treated by radical surgery or radiotherapy. However, this type of treatment is associated with urinary, sexual, and bowel dysfunction. The severity and frequency of these adverse effects differ between treatments (Wilt et al.,
2008). When tumors disseminate, androgen deprivation therapy is adopted, since cells will not grow and survive without androgens. The standard procedure of androgen deprivation (AD) by surgical or chemical castration usually results in incomplete tumor regression and the tumor eventually becomes castration-independent. This can be explained, at least in part, because AD induces a senescent-like phenotype in a subset of androgen-sensitive cancer cells (Ewald et al.,
2013).
Recent advances in PCa treatment culminated in the approval of Firmagon (degarelix), Jevtana (cabazitaxel), Provenge (sipuleucel-T), and Zytiga (abiraterone acetate) (US Food and Drug Administration,
2010; European Medicines Agency,
2009;
2011a;
2011b). Degarelix is a gonadotrophin-releasing hormone antagonist, which reduces the amount of T, and it is indicated for the hormone-sensitive stage of PCa (European Medicines Agency,
2009; Oudard,
2013). Sipuleucel-T has been a remarkable development as it was the first FDA-approved vaccine for PCa treatment, and it is currently used to treat androgen-independent (non-metastatic or minimally metastatic) PCa (Oudard,
2013). Despite the improved survival in patients with metastatic castration-resistant PCa, sipuleucel-T lacks accurate biomarkers to assess the response to immunotherapy (Kim et al.,
2012). In more advanced symptomatic metastatic stages, cabazitaxel or abiraterone acetate is commonly applied (Oudard,
2013). The optimism of patients is increasing with these new products for PCa treatment. However, overall survival remains relatively short (Antonarakis and Eisenberger,
2011).
Simultaneous with progress in the comprehension of PCa genetics and the molecular pathways involved, new therapeutic targets have been investigated, especially for advanced and metastatic cancers. Manipulation of cell signaling pathways, targeting of specific protein interaction genes, antisense therapy, anti-angiogenic and pro-apoptotic therapies are now challenges for biomedical research.
Several attempts have been made to eradicate advanced metastatic and hormone-refractory PCa mainly through targeting of AR signaling (Leibowitz-Amit and Joshua,
2012). Apparently, AR targeting via ASC-J9, an AR degradation inducer, leads to tumor suppression through the induction of autophagy (Jiang et al.,
2012). Other studies have been focused on the PI3K/Akt/mTOR pathway, testing inhibitors for each kinase. Inhibitors of mTOR are most promising, and phases I and II clinical trials are already underway (Morgan et al.,
2009). The verification that loss of PTEN expression results in a down-regulation of CXCR4-mediated events and in the subsequent activation of PI3K/AKT and ERK1/2, also drew attention to the promising antagonistic effect of this receptor (Chetram et al.,
2011).
Despite the attention given to kinases as therapeutic targets, efforts are now being made by pharmaceutical companies to investigate the role of phosphatases. Nevertheless, the only FDA-approved drugs targeting a protein phosphatase are cyclosporine A and FK506, immunosuppressors that inhibit phospho-protein phosphatase 3 (PPP3) (or calcineurin). Because of the numerous functions of PPP1 and PPP2 catalytic subunits, the long-term usage of these enzyme inhibitors is associated with nephrotoxicity and hepatotoxicity. For this reason, a more satisfactory option seems to be to target PPP1 interacting proteins (PIPs) instead of protein phosphatases directly, as they are more event, tissue, and subcellular compartment specific (Fardilha et al.,
2010). Two targeted PPP1-PIP complexes have already been described: (1) the PPP1-GADD34 complex, which is diminished in cells treated with salubrinal—a small molecule that protects the cell from ER-stress-induced apoptosis; and (2) PPP1 and histone deacetylases (HDACs), an example of which is trichostatin A, which disrupt the PPP1-HDAC6 complex in glioblastoma and PCa cells (McConnell and Wadzinski,
2009).
Other potential therapeutic agents are miRNAs. Either by anti-sense oligonucleotide inactivation of oncogenic miRNAs or by restoration of down-regulated mRNAs, it is possible to inhibit tumor growth, decrease lung metastases, and extend survival in mice (Vandenboom Ii et al.,
2008; Gordanpour et al.,
2012). A recent study proved that miR-185 could function as a tumor suppressor gene by targeting AR signaling (Qu et al.,
2013).
6. Future directions
Despite the recent positive progress observed in PCa incidence and mortality rates, efforts have to be made to achieve a better understanding of PCa precancerous lesions and of the factors triggering PCa development. This comprehension would enable a more proactive action against PCa progression. Emerging biomarkers need validation to improve PCa management and to reduce population differences in access to accurate diagnostic and prognostic procedures. The exploration of molecular targets and interactions for PCa treatment has been surprisingly rewarding and promising, with several benefits achieved in blocking progression and causing regression of metastases.
* Project supported by Fundação para a Ciência e Tecnologia (FCT) (PTDC/QUI-BIQ/118492/2010) and Fundo Europeu de Desenvolvimento Regional (FEDER) (FCOMP-01-0124-FEDER-020895), PortugalCompliance with ethics guidelines Juliana FELGUEIRAS, Joana Vieira SILVA, and Margarida FARDILHA declare that they have no conflict of interest.References
[1] Aaronson, D.S., Muller, M., Neves, S.R., 2007. An androgen-IL-6-Stat3 autocrine loop re-routes EGF signal in prostate cancer cells.
Mol Cell Endocrinol, 270(1-2):50-56.


[2] Abate-Shen, C., Shen, M.M., 2000. Molecular genetics of prostate cancer.
Genes Develop, 14(19):2410-2434.
[3] Abdulghani, J., Gu, L., Dagvadorj, A., 2008. Stat3 promotes metastatic progression of prostate cancer.
Am J Pathol, 172(6):1717-1728.
[4] Alaoui-Jamali, M.A., Xu, Y.J., 2006. Proteomic technology for biomarker profiling in cancer: an update.
J Zhejiang Univ-Sci B, 7(6):411-420.
[5] Al-Azayzih, A., Gao, F., Goc, A., 2012. TGFβ1 induces apoptosis in invasive prostate cancer and bladder cancer cells via Akt-independent, p38 MAPK and JNK/SAPK-mediated activation of caspases.
Biochem Biophys Res Commun, 427(1):165-170.
[6] Al-Maghrebi, M., Kehinde, E.O., Anim, J.T., 2012. The role of combined measurement of tissue mRNA levels of AMACR and survivin in the diagnosis and risk stratification of patients with suspected prostate cancer.
Int Urol Nephrol, 44(6):1681-1689.
[7] Almasi, C.E., Brasso, K., Iversen, P., 2011. Prognostic and predictive value of intact and cleaved forms of the urokinase plasminogen activator receptor in metastatic prostate cancer.
Prostate, 71(8):899-907.
[8] Amin, M.M., Jeyaganth, S., Fahmy, N., 2007. Subsequent prostate cancer detection in patients with prostatic intraepithelial neoplasia or atypical small acinar proliferation.
Can Urol Assoc J, 1(3):245-249.
[9] Antonarakis, E.S., Eisenberger, M.A., 2011. Expanding treatment options for metastatic prostate cancer.
N Engl J Med, 364(21):2055-2058.
[10] Antonarakis, E.S., Keizman, D., Zhang, Z., 2012. An immunohistochemical signature comprising PTEN, MYC, and Ki67 predicts progression in prostate cancer patients receiving adjuvant docetaxel after prostatectomy.
Cancer, 118(24):6063-6071.
[11] Azevedo, A., Cunha, V., Teixeira, A.L., 2011. IL-6/IL-6R as a potential key signaling pathway in prostate cancer development.
World J Clin Oncol, 2(12):384-396.
[12] Barrack, E.R., 1997. TGFβ in prostate cancer: a growth inhibitor that can enhance tumorigenicity.
Prostate, 31(1):61-70.
[13] Battisti, V., Maders, L.D., Bagatini, M.D., 2011. Oxidative stress and antioxidant status in prostate cancer patients: relation to Gleason score, treatment and bone metastasis.
Biomed Pharmacother, 65(7):516-524.
[14] Bellacosa, A., Larue, L., 2010. PI3K/AKT pathway and the epithelial-mesenchymal transition. Cancer Genome and Tumor Microenvironment. Springer,New York :11-32.
[15] Bello-DeOcampo, D., Tindall, D.J., 2003. TGF-β/Smad signaling in prostate cancer.
Curr Drug Targets, 4(3):197-207.
[16] Bernardini, S., Miano, R., Iori, R., 2004. Hypermethylation of the CpG islands in the promoter region of the
GSTP1 gene in prostate cancer: a useful diagnostic and prognostic marker?.
Clin Chim Acta, 350(1-2):181-188.
[17] Bhardwaj, A., Singh, S., Srivastava, S.K., 2011. Modulation of protein phosphatase 2A activity alters androgen-independent growth of prostate cancer cells: therapeutic implications.
Mol Cancer Ther, 10(5):720-731.
[18] Bickers, B., Aukim-Hastie, C., 2009. New molecular biomarkers for the prognosis and management of prostate cancer—the post PSA era.
Anticancer Res, 29(8):3289-3298.
[19] Bonkhoff, H., 1998. Analytical molecular pathology of epithelial-stromal interactions in the normal and neoplastic prostate.
Anal Quant Cytol Histol, 20(5):437-442.
[20] Bonkhoff, H., Fixemer, T., Hunsicker, I., 1999. Estrogen receptor expression in prostate cancer and premalignant prostatic lesions.
Am J Pathol, 155(2):641-647.
[21] Borley, N., Feneley, M.R., 2008. Prostate cancer: diagnosis and staging.
Asian J Androl, 11(1):74-80.
[22] Bostwick, D.G., 1989. The pathology of early prostate cancer.
CA Cancer J Clin, 39(6):376-393.
[23] Bostwick, D.G., Brawer, M.K., 1987. Prostatic intra-epithelial neoplasia and early invasion in prostate cancer.
Cancer, 59(4):788-794.
[24] Bostwick, D.G., Pacelli, A., Lopez-Beltran, A., 1996. Molecular biology of prostatic intraepithelial neoplasia.
Prostate, 29(2):117-134.
[25] Boyd, L.K., Mao, X., Lu, Y.J., 2012. The complexity of prostate cancer: genomic alterations and heterogeneity.
Nat Rev Urol, 9(11):652-664.
[26] Brand, T.C., Bermejo, C., Canby-Hagino, E., 2008. Association of polymorphisms in TGFB1 and prostate cancer prognosis.
J Urol, 179(2):754-758.
[27] Brodin, G., Dijke, P.T., Funa, K., 1999. Increased Smad expression and activation are associated with apoptosis in normal and malignant prostate after castration.
Cancer Res, 59(11):2731-2738.
[28] Bryzgunova, O.E., Morozkin, E.S., Yarmoschuk, S.V., 2008. Methylation-specific sequencing of GSTP1 gene promoter in circulating/extracellular DNA from blood and urine of healthy donors and prostate cancer patients.
Ann N Y Acad Sci, 1137(1):222-225.
[29] Bubendorf, L., Schopfer, A., Wagner, U., 2000. Metastatic patterns of prostate cancer: an autopsy study of 1589 patients.
Hum Pathol, 31(5):578-583.
[30] Buchanan, G., Greenberg, N.M., Scher, H.I., 2001. Collocation of androgen receptor gene mutations in prostate cancer.
Clin Cancer Res, 7(5):1273-1281.
[31] Bussemakers, M.J., van Bokhoven, A., Verhaegh, G.W., 1999. DD3: a new prostate-specific gene, highly overexpressed in prostate cancer.
Cancer Res, 59(23):5975-5979.
[32] Carruba, G., 2007. Estrogen and prostate cancer: an eclipsed truth in an androgen-dominated scenario.
J Cell Biochem, 102(4):899-911.
[33] Cavalieri, E.L., Rogan, E.G., 2006. A unified mechanism in the initiation of cancer.
Ann N Y Acad Sci, 959:341-354.
[34] Chan, S.C., Li, Y.M., Dehm, S.M., 2012. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal.
J Biol Chem, 287(23):19736-19749.
[35] Chen, C.L., Mahalingam, D., Osmulski, P., 2013. Single-cell analysis of circulating tumor cells identifies cumulative expression patterns of EMT-related genes in metastatic prostate cancer.
Prostate, 73(8):813-826.
[36] Chen, J., De, S., Brainard, J., 2004. Metastatic properties of prostate cancer cells are controlled by VEGF.
Cell Commun Adhes, 11(1):1-11.
[37] Chen, S., Kesler, C.T., Paschal, B.M., 2009. Androgen receptor phosphorylation and activity are regulated by an association with protein phosphatase 1.
J Biol Chem, 284(38):25576-25584.
[38] Chen, Y., Li, J., Yu, X., 2013. APC gene hypermethylation and prostate cancer: a systematic review and meta-analysis.
Eur J Hum Genet, 21(9):929-935.
[39] Chen, Z., Trotman, L.C., Shaffer, D., 2005. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis.
Nature, 436(7051):725-730.
[40] Chetram, M.A., Odero-Marah, V., Hinton, C.V., 2011. Loss of PTEN permits CXCR4-mediated tumorigenesis through ERK1/2 in prostate cancer cells.
Mol Cancer Res, 9(1):90-102.
[41] Choucair, K., Ejdelman, J., Brimo, F., 2012. PTEN genomic deletion predicts prostate cancer recurrence and is associated with low AR expression and transcriptional activity.
BMC Cancer, 12(1):543
[42] Clarke, N.W., Hart, C.A., Brown, M.D., 2009. Molecular mechanisms of metastasis in prostate cancer.
Asian J Androl, 11(1):57-67.
[43] Coleman, K.M., Smith, C.L., 2001. Intracellular signaling pathways: nongenomic actions of estrogens and ligand-independent activation of estrogen receptors.
Front Biosci, 6(1):D1379-D1391.
[44] Committee on Developing Biomarker-Based Tools for Cancer Screening, Diagnosis, and Treatment, 2007. Methods, tools, and resources needed to discover and develop biomarkers. Cancer Biomarkers: The Promises and Challenges of Improving Detection and Treatment. National Academies Press,Washington, DC :
[45] Craft, C.S., Romero, D., Vary, C.P., 2007. Endoglin inhibits prostate cancer motility via activation of the ALK2-Smad1 pathway.
Oncogene, 26(51):7240-7250.
[46] Craft, N., Shostak, Y., Carey, M., 1999. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase.
Nat Med, 5(3):280-285.
[47] Crawford, E.D., 2009. Understanding the epidemiology, natural history, and key pathways involved in prostate cancer.
Urology, 73(5 Suppl.):S4-S10.
[48] Crawford, E.D., Rove, K.O., Trabulsi, E.J., 2012. Diagnostic performance of PCA3 to detect prostate cancer in men with increased prostate specific antigen: a prospective study of 1962 cases.
J Urol, 188(5):1726-1731.
[49] Dai, J., Keller, J., Zhang, J., 2005. Bone morphogenetic protein-6 promotes osteoblastic prostate cancer bone metastases through a dual mechanism.
Cancer Res, 65(18):8274-8285.
[50] Danila, D.C., Anand, A., Sung, C.C., 2011. TMPRSS2-ERG status in circulating tumor cells as a predictive biomarker of sensitivity in castration-resistant prostate cancer patients treated with abiraterone acetate.
Eur Urol, 60(5):897-904.
[51] Darby, S., Cross, S.S., Brown, N.J., 2008. BMP-6 over-expression in prostate cancer is associated with increased Id-1 protein and a more invasive phenotype.
J Pathol, 214(3):394-404.
[52] Darson, M.F., Pacelli, A., Roche, P., 1997. Human glandular kallikrein 2 (hK2) expression in prostatic intraepithelial neoplasia and adenocarcinoma: a novel prostate cancer marker.
Urology, 49(6):857-862.
[53] Davidsson, S., Fiorentino, M., Andren, O., 2011. Inflammation, focal atrophic lesions, and prostatic intraepithelial neoplasia with respect to risk of lethal prostate cancer.
Cancer Epidemiol Biomarkers Prev, 20(10):2280-2287.
[54] de Marzo, A.M., Marchi, V.L., Epstein, J.I., 1999. Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis.
Am J Pathol, 155(6):1985-1992.
[55] Derynck, R., Feng, X.H., 1997. TGF-β receptor signaling.
Biochim Biophys Acta, 1333(2):F105-F150.
[56] Djulbegovic, M., Beyth, R.J., Neuberger, M.M., 2010. Screening for prostate cancer: systematic review and meta-analysis of randomised controlled trials.
BMJ, 14(341):c4543
[57] Donaldson, L., Fordyce, C., Gilliland, F., 1999. Association between outcome and telomere DNA content in prostate cancer.
J Urol, 162(5):1788-1792.
[58] Drudge-Coates, L., Turner, B., 2012. Prostate cancer overview. Part 2: metastatic prostate cancer.
Brit J Nurs, 21(18):S23-S24.
[59] Dubrovska, A., Kim, S., Salamone, R.J., 2009. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations.
PNAS, 106(1):268-273.
[60] Edlund, S., Bu, S., Schuster, N., 2003. Transforming growth factor-β1 (TGF-β)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 by TGF-β-activated kinase 1 and mitogen-activated protein kinase kinase 3.
Mol Biol Cell, 14(2):529-544.
[61] Ellem, S.J., Schmitt, J.F., Pedersen, J.S., 2004. Local aromatase expression in human prostate is altered in malignancy.
J Clin Endocrinol Metab, 89(5):2434-2441.
[62] European Medicines Agency, 2009.
Firmagon, Available from
,:
[63] European Medicines Agency, 2011.
Jevtana, Available from
,:
[64] European Medicines Agency, 2011.
Zytiga, Available from
,:
[65] Ewald, J.A., Desotelle, J.A., Church, D.R., 2013. Androgen deprivation induces senescence characteristics in prostate cancer cells
in vitro and
in vivo
.
Prostate, 73(4):337-345.
[66] Fardilha, M., Esteves, S.L.C., Korrodi-Gregorio, L., 2010. The physiological relevance of protein phosphatase 1 and its interacting proteins to health and disease.
Curr Med Chem, 17(33):3996-4017.
[67] Fardilha, M., Esteves, S.L., Korrodi-Gregorio, L., 2011. Identification of the human testis protein phosphatase 1 interactome.
Biochem Pharmacol, 82(10):1403-1415.
[68] Faria, P.C., Saba, K., Neves, A.F., 2007. Transforming growth factor-beta 1 gene polymorphisms and expression in the blood of prostate cancer patients.
Cancer Invest, 25(8):726-732.
[69] Feldman, B.J., Feldman, D., 2001. The development of androgen-independent prostate cancer.
Nat Rev Cancer, 1(1):34-45.
[70] Feng, S., Dakhova, O., Creighton, C.J., 2013. The endocrine fibroblast growth factor FGF19 promotes prostate cancer progression.
Cancer Res, 73(8):2551-2562.
[71] Ferlay, J., Parkin, D.M., Steliarova-Foucher, E., 2010. Estimates of cancer incidence and mortality in Europe in 2008.
Eur J Cancer, 46(4):765-781.
[72] Ferreira, L.B., Palumbo, A., de Mello, K.D., 2012. PCA3 noncoding RNA is involved in the control of prostate-cancer cell survival and modulates androgen receptor signaling.
BMC Cancer, 12:507
[73] Ferrer, F.A., Miller, L.J., Andrawis, R.I., 1997. Vascular endothelial growth factor (VEGF) expression in human prostate cancer: in situ and
in vitro expression of VEGF by human prostate cancer cells.
J Urol, 157(6):2329-2333.
[74] Festuccia, C., Bologna, M., Gravina, G.L., 1999. Osteoblast conditioned media contain TGF-β1 and modulate the migration of prostate tumor cells and their interactions with extracellular matrix components.
Int J Cancer, 81(3):395-403.
[75] Fisher, G., Yang, Z.H., Kudahetti, S., 2013. Prognostic value of Ki-67 for prostate cancer death in a conservatively managed cohort.
Brit J Cancer, 108(2):271-277.
[76] Fixemer, T., Remberger, K., Bonkhoff, H., 2003. Differential expression of the estrogen receptor beta (ERβ) in human prostate tissue, premalignant changes, and in primary, metastatic, and recurrent prostatic adenocarcinoma.
Prostate, 54(2):79-87.
[77] Fordyce, C.A., Heaphy, C.M., Joste, N.E., 2005. Association between cancer-free survival and telomere DNA content in prostate tumors.
J Urol, 173(2):610-614.
[78] Foster, C.S., Bostwick, D.G., Bonkhoff, H., 2000. Cellular and molecular pathology of prostate cancer precursors.
Scand J Urol Nephrol Suppl, 34(1):19-43.
[79] Gabel, S., Benefield, J., Meisinger, J., 1999. Protein phosphatases 1 and 2A maintain endothelial cells in a resting state, limiting the motility that is needed for the morphogenic process of angiogenesis.
Otolaryngol Head Neck Surg, 121(4):463-468.
[80] Gao, J., Arnold, J.T., Isaacs, J.T., 2001. Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells.
Cancer Res, 61(13):5038-5044.
[81] Gao, N., Zhang, Z., Jiang, B.H., 2003. Role of PI3K/AKT/mTOR signaling in the cell cycle progression of human prostate cancer.
Biochem Biophys Res Commun, 310(4):1124-1132.
[82] Gao, X., Zacharek, A., Grignon, D., 1995. High-frequency of loss of expression and allelic deletion of the apc and mcc genes in human prostate-cancer.
Int J Oncol, 6(1):111-117.
[83] Garnero, P., 2001. Markers of bone turnover in prostate cancer.
Cancer Treat Rev, Discussion 193-186,27(3):187-192.
[84] Garnero, P., Buchs, N., Zekri, J., 2000. Markers of bone turnover for the management of patients with bone metastases from prostate cancer.
Brit J Cancer, 82(4):858-864.
[85] Gnanapragasam, V.J., Robson, C.N., Neal, D.E., 2002. Regulation of FGF8 expression by the androgen receptor in human prostate cancer.
Oncogene, 21(33):5069-5080.
[86] Goc, A., Al-Husein, B., Kochuparambil, S.T., 2011. PI3 kinase integrates Akt and MAP kinase signaling pathways in the regulation of prostate cancer.
Int J Oncol, 38(1):267-277.
[87] Goel, M.M., Agrawal, D., Natu, S.M., 2011. Hepsin immunohistochemical expression in prostate cancer in relation to Gleason’s grade and serum prostate specific antigen.
Indian J Pathol Microbiol, 54(3):476-481.
[88] Goering, W., Kloth, M., Schulz, W.A., 2012. DNA methylation changes in prostate cancer.
Methods Mol Biol, 863:47-66.
[89] Gordanpour, A., Nam, R.K., Sugar, L., 2012. MicroRNAs in prostate cancer: from biomarkers to molecularly-based therapeutics.
Prostate Cancer Prostatic Dis, 15(4):314-319.
[90] Grignon, D.J., 2004. Unusual subtypes of prostate cancer.
Modern Pathol, 17(3):316-327.
[91] Gu, L., Dagvadorj, A., Lutz, J., 2010. Transcription factor Stat3 stimulates metastatic behavior of human prostate cancer cells
in vivo, whereas Stat5b has a preferential role in the promotion of prostate cancer cell viability and tumor growth.
Am J Pathol, 176(4):1959-1972.
[92] Gueron, G., de Siervi, A., Vazquez, E., 2012. Advanced prostate cancer: reinforcing the strings between inflammation and the metastatic behavior.
Prostate Cancer Prostatic Dis, 15(3):213-221.
[93] Guo, Y., Jacobs, S.C., Kyprianou, N., 1997. Down-regulation of protein and mRNA expression for transforming growth factor-β (TGF-β1) type I and type II receptors in human prostate cancer.
Int J Cancer, 71(4):573-579.
[94] Guo, Z., Yang, X., Sun, F., 2009. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth.
Cancer Res, 69(6):2305-2313.
[95] Gupta-Elera, G., Garrett, A.R., Robison, R.A., 2012. The role of oxidative stress in prostate cancer.
Eur J Cancer Prev, 21(2):155-162.
[96] Haas, G.P., Delongchamps, N.B., Jones, R.F., 2007. Needle biopsies on autopsy prostates: sensitivity of cancer detection based on true prevalence.
J Natl Cancer Inst, 99(19):1484-1489.
[97] Hamdy, F.C., Autzen, P., Robinson, M.C., 1997. Immunolocalization and messenger RNA expression of bone morphogenetic protein-6 in human benign and malignant prostatic tissue.
Cancer Res, 57(19):4427-4431.
[98] Hayes, S.A., Huang, X., Kambhampati, S., 2003. p38 MAP kinase modulates Smad-dependent changes in human prostate cell adhesion.
Oncogene, 22(31):4841-4850.
[99] He, H.C., Bi, X.C., Zheng, Z.W., 2009. Real-time quantitative RT-PCR assessment of PIM-1 and hK2 mRNA expression in benign prostate hyperplasia and prostate cancer.
Med Oncol, 26(3):303-308.
[100] Heemers, H.V., Tindall, D.J., 2007. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex.
Endocrine Rev, 28(7):778-808.
[101] Hegele, A., Wahl, H.G., Varga, Z., 2007. Biochemical markers of bone turnover in patients with localized and metastasized prostate cancer.
BJU Int, 99(2):330-334.
[103] Heinlein, C.A., Chang, C., 2004. Androgen receptor in prostate cancer.
Endocrine Rev, 25(2):276-308.
[104] Hrnberg, E., Ylitalo, E.B., Crnalic, S., 2011. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival.
PLoS ONE, 6(4):e19059
[105] Horvath, L.G., Henshall, S.M., Lee, C.S., 2001. Frequent loss of estrogen receptor-β expression in prostate cancer.
Cancer Res, 61(14):5331-5335.
[106] Horvath, L.G., Henshall, S.M., Kench, J.G., 2004. Loss of BMP2, Smad8, and Smad4 expression in prostate cancer progression.
Prostate, 59(3):234-242.
[107] Huggins, C., Hodges, C.V., 1972. Studies on prostatic cancer: I. The effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate.
CA Cancer J Clin, 22(4):232-240.
[108] Ilic, D., OConnor, D., Green, S., 2011. Screening for prostate cancer: an updated Cochrane systematic review.
BJU Int, 107(6):882-891.
[109] Jariwala, U., Prescott, J., Jia, L., 2007. Identification of novel androgen receptor target genes in prostate cancer.
Mol Cancer, 6(1):1-15.
[110] Jemal, A., Bray, F., Center, M.M., 2011. Global cancer statistics.
CA Cancer J Clin, 61(2):69-90.
[111] Jennbacken, K., Vallbo, C., Wang, W., 2005. Expression of vascular endothelial growth factor C (VEGF-C) and VEGF receptor-3 in human prostate cancer is associated with regional lymph node metastasis.
Prostate, 65(2):110-116.
[112] Jiang, Q., Yeh, S., Wang, X., 2012. Targeting androgen receptor leads to suppression of prostate cancer via induction of autophagy.
J Urol, 188(4):1361-1368.
[113] Josefsson, A., Wikstrom, P., Egevad, L., 2012. Low endoglin vascular density and Ki67 index in Gleason score 6 tumours may identify prostate cancer patients suitable for surveillance.
Scand J Urol Nephrol, 46(4):247-257.
[114] Kageyama, Y., Kamata, S., Yonese, J., 1997. Telomere length and telomerase activity in bladder and prostate cancer cell lines.
Int J Urol, 4(4):407-410.
[115] Kang, H.Y., Lin, H.K., Hu, Y.C., 2001. From transforming growth factor-beta signaling to androgen action: identification of Smad3 as an androgen receptor coregulator in prostate cancer cells.
PNAS, 98(6):3018-3023.
[116] Kantoff, P.W., Higano, C.S., Shore, N.D., 2010. Sipuleucel-T immunotherapy for castration-resistant prostate cancer.
N Engl J Med, 363(5):411-422.
[117] Karam, J.A., Svatek, R.S., Karakiewicz, P.I., 2008. Use of preoperative plasma endoglin for prediction of lymph node metastasis in patients with clinically localized prostate cancer.
Clin Cancer Res, 14(5):1418-1422.
[118] Kazma, R., Mefford, J.A., Cheng, I., 2012. Association of the innate immunity and inflammation pathway with advanced prostate cancer risk.
PLoS ONE, 7(12):e51680
[119] Kelly, K.A., Setlur, S.R., Ross, R., 2008. Detection of early prostate cancer using a hepsin-targeted imaging agent.
Cancer Res, 68(7):2286-2291.
[120] Kim, I.Y., Ahn, H.J., Lang, S., 1998. Loss of expression of transforming growth factor-β receptors is associated with poor prognosis in prostate cancer patients.
Clin Cancer Res, 4(7):1625-1630.
[121] Kim, J., Bilusic, M., Heery, C., 2012. Therapeutic cancer vaccines in prostate cancer: the quest for intermediate markers of response.
Cancers, 4(4):1229-1246.
[122] Klezovitch, O., Chevillet, J., Mirosevich, J., 2004. Hepsin promotes prostate cancer progression and metastasis.
Cancer Cell, 6(2):185-195.
[123] Knudsen, B.S., Vasioukhin, V., 2010. Mechanisms of prostate cancer initiation and progression. Advances in Cancer Research. Academic Press,United States of America :
[124] Koca, O., Caliskan, S., Ozturk, M.I., 2011. Significance of atypical small acinar proliferation and high-grade prostatic intraepithelial neoplasia in prostate biopsy.
Korean J Urol, 52(11):736-740.
[125] Korrodi-Gregorio, L., Teixeira, A.L., Medeiros, R., 2012. TGFβ pathway. Essentials of Cell Signaling. (in Portuguese), Afrontamento,Aveiro :199-234.
[126] Kuiper, G.G., Enmark, E., Pelto-Huikko, M., 1996. Cloning of a novel receptor expressed in rat prostate and ovary.
PNAS, 93(12):5925-5930.
[127] Kumano, M., Miyake, H., Muramaki, M., 2009. Expression of urokinase-type plasminogen activator system in prostate cancer: correlation with clinicopathological outcomes in patients undergoing radical prostatectomy.
Urol Oncol, 27(2):180-186.
[128] Lakshman, M., Huang, X., Ananthanarayanan, V., 2011. Endoglin suppresses human prostate cancer metastasis.
Clin Exp Metast, 28(1):39-53.
[129] Landstrom, M., Heldin, N.E., Bu, S., 2000. Smad7 mediates apoptosis induced by transforming growth factor β in prostatic carcinoma cells.
Curr Biol, 10(9):535-538.
[130] Langenfeld, E.M., Langenfeld, J., 2004. Bone morphogenetic protein-2 stimulates angiogenesis in developing tumors11NIH K22 grant CA91919-01A1 and UMDNJ foundation to J. Langenfeld.
Mol Cancer Res, 2(3):141-149.
[131] Lee, C., Prins, G.S., Henneberry, M.O., 1981. Effect of estradiol on the rat prostate in the presence and absence of testosterone and pituitary.
J Androl, 2(6):293-299.
[132] Lee, C., Sintich, S.M., Mathews, E.P., 1999. Transforming growth factor-β in benign and malignant prostate.
Prostate, 39(4):285-290.
[133] Leiblich, A., Cross, S.S., Catto, J.W., 2006. Lactate dehydrogenase-B is silenced by promoter hypermethy-lation in human prostate cancer.
Oncogene, 25(20):2953-2960.
[134] Leibowitz-Amit, R., Joshua, A.M., 2012. Targeting the androgen receptor in the management of castration-resistant prostate cancer: rationale, progress, and future directions.
Curr Oncol, 19(Suppl. 3):S22-S31.
[135] Leung, Y.K., Lam, H.M., Wu, S., 2010. Estrogen receptor β2 and β5 are associated with poor prognosis in prostate cancer, and promote cancer cell migration and invasion.
Endocr Relat Cancer, 17(3):675-689.
[136] Leung, Y.K., Lee, M.T., Wang, J., 2010. Post-transcriptional regulation of estrogen receptor β isoforms in prostate cancer.
, ENDO2010 92nd Annual Meeting, San Diego, California, :
[137] Li, L., Ren, C.H., Tahir, S.A., 2003. Caveolin-1 maintains activated Akt in prostate cancer cells through scaffolding domain binding site interactions with and inhibition of serine/threonine protein phosphatases PP1 and PP2A.
Mol Cell Biol, 23(24):9389-9404.
[138] Li, L., Ittmann, M.M., Ayala, G., 2005. The emerging role of the PI3-K-Akt pathway in prostate cancer progression.
Prostate Cancer Prostatic Dis, 8(2):108-118.
[139] Li, W., Wang, X., Li, B., 2012. Diagnostic significance of overexpression of Golgi membrane protein 1 in prostate cancer.
Urology, 80(4):952.e1-952.e7.
[140] Lin, H.K., Chen, Z., Wang, G., 2010. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence.
Nature, 464(7287):374-379.
[141] Liu, A.J., Furusato, B., Ravindranath, L., 2007. Quantitative analysis of a panel of gene expression in prostate cancer—with emphasis on NPY expression analysis.
J Zhejiang Univ-Sci B, 8(12):853-859.
[142] Liu, L., Kron, K.J., Pethe, V.V., 2011. Association of tissue promoter methylation levels of APC, TGFβ2, HOXD3 and RASSF1A with prostate cancer progression.
Int J Cancer, 129(10):2454-2462.
[143] Liwei, L., Chunyu, L., Jie, L., 2011. Association between fibroblast growth factor receptor-4 gene polymorphism and risk of prostate cancer: a meta-analysis.
Urol Int, 87(2):159-164.
[144] Lockett, K.L., Hall, M.C., Clark, P.E., 2006. DNA damage levels in prostate cancer cases and controls.
Carcinogenesis, 27(6):1187-1193.
[145] Lonergan, P.E., Tindall, D.J., 2011. Androgen receptor signaling in prostate cancer development and progression.
J Carcinogen, 10:20
[146] Lopez, J.I., 2007. Prostate adenocarcinoma detected after high-grade prostatic intraepithelial neoplasia or atypical small acinar proliferation.
BJU Int, 100(6):1272-1276.
[147] Lotan, T.L., Gurel, B., Sutcliffe, S., 2011. PTEN protein loss by immunostaining: analytic validation and prognostic indicator for a high risk surgical cohort of prostate cancer patients.
Clin Cancer Res, 17(20):6563-6573.
[148] Lu, T.L., Huang, Y.F., You, L.R., 2013. Conditionally ablated
Pten in prostate basal cells promotes basal-to-luminal differentiation and causes invasive prostate cancer in mice.
Am J Pathol, 182(3):975-991.
[149] Lumen, N., Fonteyne, V., de Meerleer, G., 2012. Screening and early diagnosis of prostate cancer: an update.
Acta Clin Belg, 67(4):270-275.
[150] Ma, Y., Cheng, Q., Ren, Z., 2012. Induction of IGF-1R expression by EGR-1 facilitates the growth of prostate cancer cells.
Cancer Lett, 317(2):150-156.
[151] Madani, S.H., Ameli, S., Khazaei, S., 2011. Frequency of Ki-67 (MIB-1) and P53 expressions among patients with prostate cancer.
Indian J Pathol Microbiol, 54(4):688-691.
[152] Majumder, P.K., Sellers, W.R., 2005. Akt-regulated pathways in prostate cancer.
Oncogene, 24(50):7465-7474.
[153] Mak, P., Leav, I., Pursell, B., 2010. ERβ impedes prostate cancer EMT by destabilizing HIF-1α and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading.
Cancer Cell, 17(4):319-332.
[154] Makarov, D.V., Loeb, S., Getzenberg, R.H., 2009. Biomarkers for prostate cancer.
Ann Rev Med, 60:139-151.
[155] Mancuso, P.A., Chabert, C., Chin, P., 2007. Prostate cancer detection in men with an initial diagnosis of atypical small acinar proliferation.
BJU Int, 99(1):49-52.
[156] Marino, M., Galluzzo, P., Ascenzi, P., 2006. Estrogen signaling multiple pathways to impact gene transcription.
Curr Genomics, 7(8):497-508.
[157] Marques, R.B., Dits, N.F., Erkens-Schulze, S., 2011. Modulation of androgen receptor signaling in hormonal therapy-resistant prostate cancer cell lines.
PLoS ONE, 6(8):e23144
[158] Martin-Granados, C., Prescott, A.R., van Dessel, N., 2012. A role for PP1/NIPP1 in steering migration of human cancer cells.
PLoS ONE, 7(7):e40769
[159] Masuda, H., Fukabori, Y., Nakano, K., 2004. Expression of bone morphogenetic protein-7 (BMP-7) in human prostate.
Prostate, 59(1):101-106.
[160] Mattie, M.D., Benz, C.C., Bowers, J., 2006. Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies.
Mol Cancer, 5:24
[161] McConnell, J.L., Wadzinski, B.E., 2009. Targeting protein serine/threonine phosphatases for drug development.
Mol Pharmacol, 75(6):1249-1261.
[162] McPherson, S.J., Hussain, S., Balanathan, P., 2010. Estrogen receptor-β activated apoptosis in benign hyperplasia and cancer of the prostate is androgen independent and TNFα mediated.
PNAS, 107(7):3123-3128.
[163] Merz, V.W., Arnold, A.M., Studer, U.E., 1994. Differential expression of transforming growth factor-β1 and β3 as well as C-FOS mRNA in normal human prostate, benign prostatic hyperplasia and prostatic cancer.
World J Urol, 12(2):96-98.
[164] Meyer, H.A., Ahrens-Fath, I., Sommer, A., 2004. Novel molecular aspects of prostate carcinogenesis.
Biomed Pharmacother, 58(1):10-16.
[165] Miller, D.C., Hafez, K.S., Stewart, A., 2003. Prostate carcinoma presentation, diagnosis, and staging: an update from the National Cancer Data Base.
Cancer, 98(6):1169-1178.
[166] Mitchell, P.S., Parkin, R.K., Kroh, E.M., 2008. Circulating microRNAs as stable blood-based markers for cancer detection.
PNAS, 105(30):10513-10518.
[167] Miyake, H., Hara, I., Kamidono, S., 2004. Oxidative DNA damage in patients with prostate cancer and its response to treatment.
J Urol, 171(4):1533-1536.
[168] Moore, C.K., Karikehalli, S., Nazeer, T., 2005. Prognostic significance of high grade prostatic intrae-pithelial neoplasia and atypical small acinar proliferation in the contemporary era.
J Urol, 173(1):70-72.
[169] Morgan, T.M., Koreckij, T.D., Corey, E., 2009. Targeted therapy for advanced prostate cancer: inhibition of the PI3K/Akt/mTOR pathway.
Curr Cancer Drug Targets, 9(2):237-249.
[170] Moschos, S.J., Mantzoros, C.S., 2002. The role of the IGF system in cancer: from basic to clinical studies and clinical applications.
Oncology, 63(4):317-332.
[171] Naruse, K., Yamada, Y., Aoki, S., 2007. Lactate dehydrogenase is a prognostic indicator for prostate cancer patients with bone metastasis.
Hinyokika Kiyo, 53(5):287-292.
[172] Nelson, W.G., de Marzo, A.M., Isaacs, W.B., 2003. Prostate cancer.
N Engl J Med, 349(4):366-381.
[173] Nieto, M., Finn, S., Loda, M., 2007. Prostate cancer: re-focusing on androgen receptor signaling.
Int J Biochem Cell Biol, 39(9):1562-1568.
[174] Nyquist, M.D., Dehm, S.M., 2013. Interplay between genomic alterations and androgen receptor signaling during prostate cancer development and progression.
Hormones Cancer, 4(2):61-69.
[175] Ohta, S., Fuse, H., Fujiuchi, Y., 2003. Clinical significance of expression of urokinase-type plasminogen activator in patients with prostate cancer.
Anticancer Res, 23(3C):2945-2950.
[176] Okino, S.T., Pookot, D., Majid, S., 2007. Chromatin changes on the GSTP1 promoter associated with its inactivation in prostate cancer.
Mol Carcinogen, 46(10):839-846.
[177] Okotie, O.T., Roehl, K.A., Han, M., 2007. Characteristics of prostate cancer detected by digital rectal examination only.
Urology, 70(6):1117-1120.
[178] Oudard, S., 2013. Progress in emerging therapies for advanced prostate cancer.
Cancer Treatment Rev, 39(3):275-289.
[179] Pace, G., Pomante, R., Vicentini, C., 2012. Hepsin in the diagnosis of prostate cancer.
Minerva Urol Nefrol, 64(2):143-148.
[180] Parsons, J.K., Gage, W.R., Nelson, W.G., 2001. p63 protein expression is rare in prostate adenocarcinoma: implications for cancer diagnosis and carcinogenesis.
Urology, 58(4):619-624.
[181] Pasder, O., Shpungin, S., Salem, Y., 2006. Downregulation of Fer induces PP1 activation and cell-cycle arrest in malignant cells.
Oncogene, 25(30):4194-4206.
[182] Peehl, D.M., 2005. Primary cell cultures as models of prostate cancer development.
Endocrine-Related Cancer, 12(1):19-47.
[183] Perttu, M.C., Martikainen, P.M., Huhtala, H.S., 2006. Altered levels of Smad2 and Smad4 are associated with human prostate carcinogenesis.
Prostate Cancer Prostatic Dis, 9(2):185-189.
[184] Pollak, M., 2008. Insulin and insulin-like growth factor signalling in neoplasia.
Nat Rev Cancer, 8(12):915-928.
[185] Pourmand, G., Ziaee, A.A., Abedi, A.R., 2007. Role of
PTEN gene in progression of prostate cancer.
Urol Oncol, 4(2):95-100.
[186] Putzi, M.J., de Marzo, A.M., 2000. Morphologic transitions between proliferative inflammatory atrophy and high-grade prostatic intraepithelial neoplasia.
Urology, 56(5):828-832.
[187] Qi, L., Robinson, W.A., Brady, B.M., 2003. Migration and invasion of human prostate cancer cells is related to expression of VEGF and its receptors.
Anticancer Res, 23(5A):3917-3922.
[188] Qu, F., Cui, X., Hong, Y., 2013. MicroRNA-185 suppresses proliferation, invasion, migration, and tumorigenicity of human prostate cancer cells through targeting androgen receptor.
Mol Cell Biochem, 377(1-2):121-130.
[189] Raaijmakers, R., Kirkels, W.J., Roobol, M.J., 2002. Complication rates and risk factors of 5802 transrectal ultrasound-guided sextant biopsies of the prostate within a population-based screening program.
Urology, 60(5):826-830.
[190] Raaijmakers, R., de Vries, S.H., Blijenberg, B.G., 2007. hK2 and free PSA, a prognostic combination in predicting minimal prostate cancer in screen-detected men within the PSA range 4–10 ng/ml.
Eur Urol, 52(5):1358-1364.
[191] Reynolds, A.R., Kyprianou, N., 2006. Growth factor signalling in prostatic growth: significance in tumour development and therapeutic targeting.
Brit J Pharmacol, 147(S2):S144-S152.
[192] Rhea, J.M., Molinaro, R.J., 2011. Cancer biomarkers: surviving the journey from bench to bedside.
Med Lab Obs, 43(3):10-18.
[193] Ricke, W.A., McPherson, S.J., Bianco, J.J., 2008. Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor alpha signaling.
FASEB J, 22(5):1512-1520.
[194] Rolan, P., 1997. The contribution of clinical pharmacology surrogates and models to drug development—a critical appraisal.
Brit J Clin Pharmacol, 44(3):219-225.
[195] Romero, D., Terzic, A., Conley, B.A., 2010. Endoglin phosphorylation by ALK2 contributes to the regulation of prostate cancer cell migration.
Carcinogenesis, 31(3):359-366.
[196] Rubin, M.A., Zhou, M., Dhanasekaran, S.M., 2002. Alpha-methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer.
JAMA, 287(13):1662-1670.
[197] Saez, C., Gonzalez-Baena, A.C., Japon, M.A., 1998. Regressive changes in finasteride-treated human hyperplastic prostates correlate with an upregulation of TGF-β receptor expression.
Prostate, 37(2):84-90.
[198] Salmena, L., Carracedo, A., Pandolfi, P.P., 2008. Tenets of PTEN tumor suppression.
Cell, 133(3):403-414.
[199] Samaratunga, H., Gardiner, R.A., Yaxley, J., 2006. Atypical prostatic glandular proliferations on needle biopsy: diagnostic implications, use of immunohistochemistry, and clinical significance.
Anal Quant Cytol Histol, 28(2):104-110.
[200] Schindl, M., Oberhuber, G., Obermair, A., 2001. Overexpression of Id-1 protein is a marker for unfavorable prognosis in early-stage cervical cancer.
Cancer Res, 61(15):5703-5706.
[201] Schindl, M., Schoppmann, S.F., Strobel, T., 2003. Level of Id-1 protein expression correlates with poor differentiation, enhanced malignant potential, and more aggressive clinical behavior of epithelial ovarian tumors.
Clin Cancer Res, 9(2):779-785.
[202] Schrder, F.H., Gosselaar, C., Roemeling, S., 2006. PSA and the detection of prostate cancer after 2005. Part I.
EAU-EBU Update Series, 4(1):2-12.
[203] Sfanos, K.S., de Marzo, A.M., 2012. Prostate cancer and inflammation: the evidence.
Histopathology, 60(1):199-215.
[204] Shi, W., Sun, C., He, B., 2004. GADD34-PP1c recruited by Smad7 dephosphorylates TGFβ type I receptor.
J Cell Biol, 164(2):291-300.
[205] Shteynshlyuger, A., Andriole, G.L., 2010. Prostate cancer: to screen or not to screen?.
Urol Clin N Am, 37(1):1-9.
[206] Singh, P.B., Matanhelia, S.S., Martin, F.L., 2008. A potential paradox in prostate adenocarcinoma progression: oestrogen as the initiating driver.
Eur J Cancer, 44(7):928-936.
[207] Smith, R.A., Mettlin, C.J., Eyre, H., 2003. Cancer screening and early detection. Holland-Frei Cancer Medicine. BC Decker,Canada :
[208] Som, A., Tu, S.M., Liu, J., 2012. Response in bone turnover markers during therapy predicts overall survival in patients with metastatic prostate cancer: analysis of three clinical trials.
Brit J Cancer, 107(9):1547-1553.
[209] Soulitzis, N., Karyotis, I., Delakas, D., 2006. Expression analysis of peptide growth factors VEGF, FGF2, TGFB1, EGF and IGF1 in prostate cancer and benign prostatic hyperplasia.
Int J Oncol, 29(2):305-314.
[210] Squire, J.A., 2009. TMPRSS2-ERG and PTEN loss in prostate cancer.
Nat Genet, 41(5):509-510.
[211] Stamey, T.A., Johnstone, I.M., McNeal, J.E., 2002. Preoperative serum prostate specific antigen levels between 2 and 22 ng/ml correlate poorly with post-radical prostatectomy cancer morphology: prostate specific antigen cure rates appear constant between 2 and 9 ng/ml.
J Urol, 167(1):103-111.
[212] Stattin, P., Rinaldi, S., Biessy, C., 2004. High levels of circulating insulin-like growth factor-I increase prostate cancer risk: a prospective study in a population-based nonscreened cohort.
J Clin Oncol, 22(15):3104-3112.
[213] Steuber, T., Vickers, A.J., Serio, A.M., 2007. Comparison of free and total forms of serum human kallikrein 2 and prostate-specific antigen for prediction of locally advanced and recurrent prostate cancer.
Clin Chem, 53(2):233-240.
[214] Szczyrba, J., Nolte, E., Wach, S., 2011. Downregulation of Sec23A protein by miRNA-375 in prostate carcinoma.
Mol Cancer Res, 9(6):791-800.
[215] Taira, J., Higashimoto, Y., 2013. Caveolin-1 interacts with protein phosphatase 5 and modulates its activity in prostate cancer cells.
Biochem Biophys Res Commun, 431(4):724-728.
[216] Teishima, J., Shoji, K., Hayashi, T., 2012. Relationship between the localization of fibroblast growth factor 9 in prostate cancer cells and postoperative recurrence.
Prostate Cancer Prostatic Dis, 15(1):8-14.
[217] Thalgott, M., Rack, B., Maurer, T., 2013. Detection of circulating tumor cells in different stages of prostate cancer.
J Cancer Res Clin Oncol, 139(5):755-763.
[218] Thara, E., Dorff, T.B., Pinski, J.K., 2011. Vaccine therapy with sipuleucel-T (Provenge) for prostate cancer.
Maturitas, 69(4):296-303.
[219] Tomlins, S.A., Rhodes, D.R., Perner, S., 2005. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer.
Science, 310(5748):644-648.
[220] Treat, E.G., Heaphy, C.M., Massie, L.W., 2010. Telomere DNA content in prostate biopsies predicts early rise in prostate-specific antigen after radical prostatectomy for prostate cancer.
Urology, 75(3):724-729.
[221] Trojan, L., Thomas, D., Knoll, T., 2004. Expression of pro-angiogenic growth factors VEGF, EGF and bFGF and their topographical relation to neovascularisation in prostate cancer.
Urol Res, 32(2):97-103.
[222] Tu, W.H., Thomas, T.Z., Masumori, N., 2003. The loss of TGF-β signaling promotes prostate cancer metastasis.
Neoplasia, 5(3):267-277.
[223] Turner, B.J., Mavandadi, S., Weiner, M.G., 2011. Association of black race with follow-up of an abnormal prostate-specific antigen test.
J Natl Med Assoc, 103(2):150-157.
[225] Vandenboom Ii, T.G., Li, Y., Philip, P.A., 2008. MicroRNA and cancer: tiny molecules with major implications.
Curr Genomics, 9(2):97-109.
[226] Vander Griend, D.J., D'Antonio, J., Gurel, B., 2010. Cell-autonomous intracellular androgen receptor signaling drives the growth of human prostate cancer initiating cells.
Prostate, 70(1):90-99.
[227] Varambally, S., Laxman, B., Mehra, R., 2008. Golgi protein GOLM1 is a tissue and urine biomarker of prostate cancer.
Neoplasia, 10(11):1285-1294.
[228] Vo, B.T., Khan, S.A., 2011. Expression of nodal and nodal receptors in prostate stem cells and prostate cancer cells: autocrine effects on cell proliferation and migration.
Prostate, 71(10):1084-1096.
[229] Wang, H., Fan, L., Wei, J., 2012. Akt mediates metastasis-associated gene 1 (MTA1) regulating the expression of E-cadherin and promoting the invasiveness of prostate cancer cells.
PLoS ONE, 7(12):e46888
[230] Wang, J., Yu, W., Cai, Y., 2008. Altered fibroblast growth factor receptor 4 stability promotes prostate cancer progression.
Neoplasia, 10(8):847-856.
[231] Wang, W., Bergh, A., Damber, J.E., 2009. Morphological transition of proliferative inflammatory atrophy to high-grade intraepithelial neoplasia and cancer in human prostate.
Prostate, 69(13):1378-1386.
[232] Wikstrom, P., Lissbrant, I.F., Stattin, P., 2002. Endoglin (CD105) is expressed on immature blood vessels and is a marker for survival in prostate cancer.
Prostate, 51(4):268-275.
[233] Wilt, T.J., Shamliyan, T., Taylor, B., 2008. Comparative effectiveness of therapies for clinically localized prostate cancer. AHRQ Comparative Effectiveness Reviews. Rockville,United States of America :
[234] Woodson, K., O'Reilly, K.J., Hanson, J.C., 2008. The usefulness of the detection of GSTP1 methylation in urine as a biomarker in the diagnosis of prostate cancer.
J Urol, Discussion 511-502,179(2):508-511.
[235] Wu, Z., McRoberts, K.S., Theodorescu, D., 2007. The role of PTEN in prostate cancer cell tropism to the bone micro-environment.
Carcinogenesis, 28(7):1393-1400.
[236] Wu, Z., He, B., He, J., 2012. Upregulation of miR-153 promotes cell proliferation via downregulation of the PTEN tumor suppressor gene in human prostate cancer.
Prostate, 73(6):596-604.
[237] Xu, T., He, K., Wang, L., 2011. Prostate tumor cells with cancer progenitor properties have high telomerase activity and are rapidly killed by telomerase interference.
Prostate, 71(13):1390-1400.
[238] Yager, J.D., 2000. Endogenous estrogens as carcinogens through metabolic activation.
J Natl Cancer Inst Monogr, 2000(27):67-73.
[239] Yaman Agaoglu, F., Kovancilar, M., Dizdar, Y., 2011. Investigation of miR-21, miR-141, and miR-221 in blood circulation of patients with prostate cancer.
Tumour Biol, 32(3):583-588.
[240] Yang, F., Strand, D.W., Rowley, D.R., 2008. Fibroblast growth factor-2 mediates transforming growth factor-β action in prostate cancer reactive stroma.
Oncogene, 27(4):450-459.
[241] Yang, Q., Fung, K.M., Day, W.V., 2005. Androgen receptor signaling is required for androgen-sensitive human prostate cancer cell proliferation and survival.
Cancer Cell Int, 5(1):8
[242] Yang, X., Guo, Z., Sun, F., 2011. Novel membrane-associated androgen receptor splice variant potentiates proliferative and survival responses in prostate cancer cells.
J Biol Chem, 286(41):36152-36160.
[243] Yap, T.A., Swanton, C., de Bono, J.S., 2012. Personalization of prostate cancer prevention and therapy: are clinically qualified biomarkers in the horizon?.
EPMA J, 3(1):3
[244] Ye, L., Lewis-Russell, J.M., Kyanaston, H.G., 2007. Bone morphogenetic proteins and their receptor signaling in prostate cancer.
Histol Histopathol, 22(10):1129-1147.
[245] Ye, L., Lewis-Russell, J.M., Kynaston, H., 2007. Endogenous bone morphogenetic protein-7 controls the motility of prostate cancer cells through regulation of bone morphogenetic protein antagonists.
J Urol, 178(3 Pt 1):1086-1091.
[246] Ye, L., Kynaston, H., Jiang, W.G., 2008. Bone morphogenetic protein-9 induces apoptosis in prostate cancer cells, the role of prostate apoptosis response-4.
Mol Cancer Res, 6(10):1594-1606.
[247] Ye, L., Kynaston, H., Jiang, W.G., 2009. Bone morphogenetic protein-10 suppresses the growth and aggressiveness of prostate cancer cells through a Smad independent pathway.
J Urol, 181(6):2749-2759.
[248] Yoon, H.Y., Kim, S.K., Kim, Y.W., 2012. Combined hypermethylation of APC and GSTP1 as a molecular marker for prostate cancer: quantitative pyrosequencing analysis.
J Biomol Screen, 17(7):987-992.
[249] Yuen, H.F., Chan, Y.P., Cheung, W.L., 2008. The prognostic significance of BMP-6 signaling in prostate cancer.
Modern Pathol, 21(12):1436-1443.
[250] Yutkin, V., Al-Zahrani, A., Williams, A., 2012. Pca3 test as an adjunct in diagnosis of prostate cancer.
J Urol, 187(4):E889-E889.
[251] Zhao, Z., Zeng, G., Zhong, W., 2010. Serum early prostate cancer antigen (EPCA) as a significant predictor of incidental prostate cancer in patients undergoing transurethral resection of the prostate for benign prostatic hyperplasia.
Prostate, 70(16):1788-1798.
[252] Zhao, Z., Ma, W., Zeng, G., 2012. Preoperative serum levels of early prostate cancer antigen (EPCA) predict prostate cancer progression in patients undergoing radical prostatectomy.
Prostate, 72(3):270-279.
[253] Zhou, X., Mao, J., Ai, J., 2012. Identification of plasma lipid biomarkers for prostate cancer by lipidomics and bioinformatics.
PLoS ONE, 7(11):e48889
[254] Zhu, M.L., Kyprianou, N., 2008. Androgen receptor and growth factor signaling cross-talk in prostate cancer cells.
Endocrine-Related Cancer, 15(4):841-849.
Open peer comments: Debate/Discuss/Question/Opinion
<1>
YI@No address<No mail>
2014-02-08 10:58:25
This is an excellent review for recent progress in the molecular pathology of prostate cancer. I believe that this article would give high impact to the wide range of fields of clinical and/or biological sciences.
MM@No address<No mail>
2014-02-08 10:56:36
In this review Authors describe the pathophysiological features of PCa focusing in the main molecular processes involved the conventional approaches to PCa diagnosis and treatment as well as the recent advances in the establishment of new molecular markers for PCa management. The review is well written interesting and well done covering hot issues on PCa.