Full Text:
![]() <3714>
<3714>
Summary: ![]() <2473>
<2473>
Suppl. Mater.: ![]()
CLC number: Q987
On-line Access: 2024-08-27
Received: 2023-10-17
Revision Accepted: 2024-05-08
Crosschecked: 2014-08-30
Cited: 8
Clicked: 8554
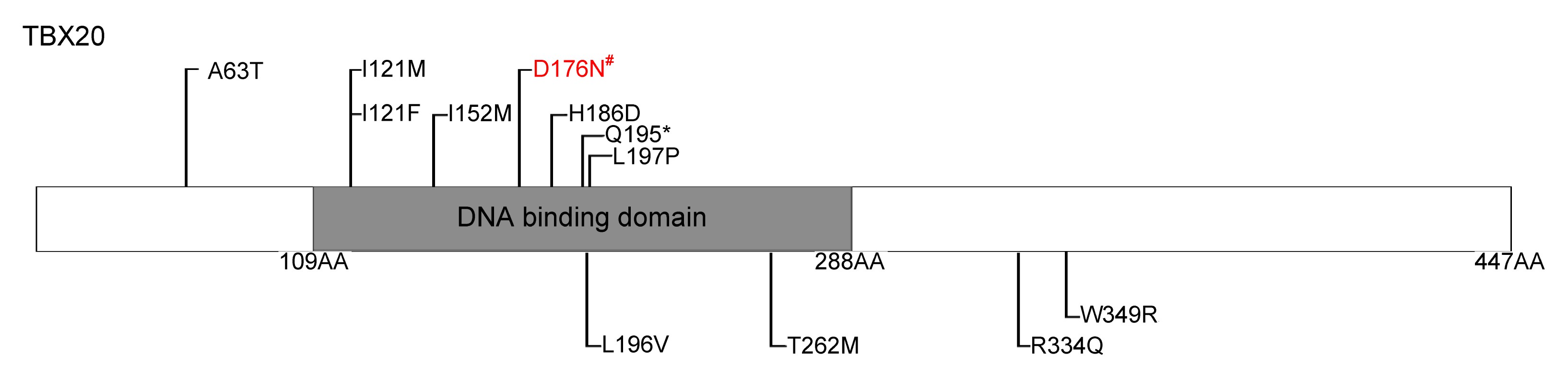
A novel variant in TBX20 (p.D176N) identified by whole-exome sequencing in combination with a congenital heart disease related gene filter is associated with familial atrial septal defect
| ||||||||||||||
Ji-jia Liu, Liang-liang Fan, Jin-lan Chen, Zhi-ping Tan, Yi-feng Yang. A novel variant in TBX20 (p.D176N) identified by whole-exome sequencing in combination with a congenital heart disease related gene filter is associated with familial atrial septal defect[J]. Journal of Zhejiang University Science B, 2014, 15(9): 830-837.
@article{title="A novel variant in TBX20 (p.D176N) identified by whole-exome sequencing in combination with a congenital heart disease related gene filter is associated with familial atrial septal defect",
author="Ji-jia Liu, Liang-liang Fan, Jin-lan Chen, Zhi-ping Tan, Yi-feng Yang",
journal="Journal of Zhejiang University Science B",
volume="15",
number="9",
pages="830-837",
year="2014",
publisher="Zhejiang University Press & Springer",
doi="10.1631/jzus.B1400062"
}
%0 Journal Article
%T A novel variant in TBX20 (p.D176N) identified by whole-exome sequencing in combination with a congenital heart disease related gene filter is associated with familial atrial septal defect
%A Ji-jia Liu
%A Liang-liang Fan
%A Jin-lan Chen
%A Zhi-ping Tan
%A Yi-feng Yang
%J Journal of Zhejiang University SCIENCE B
%V 15
%N 9
%P 830-837
%@ 1673-1581
%D 2014
%I Zhejiang University Press & Springer
%DOI 10.1631/jzus.B1400062
TY - JOUR
T1 - A novel variant in TBX20 (p.D176N) identified by whole-exome sequencing in combination with a congenital heart disease related gene filter is associated with familial atrial septal defect
A1 - Ji-jia Liu
A1 - Liang-liang Fan
A1 - Jin-lan Chen
A1 - Zhi-ping Tan
A1 - Yi-feng Yang
J0 - Journal of Zhejiang University Science B
VL - 15
IS - 9
SP - 830
EP - 837
%@ 1673-1581
Y1 - 2014
PB - Zhejiang University Press & Springer
ER -
DOI - 10.1631/jzus.B1400062
Abstract: congenital heart disease (CHD) is the leading cause of birth defects, and its etiology is not completely understood. atrial septal defect (ASD) is one of the most common defects of CHD. Previous studies have demonstrated that mutations in the transcription factor T-box 20 (TBX20) contribute to congenital ASD. whole-exome sequencing in combination with a CHD-related gene filter was used to detect a family of three generations with ASD. A novel TBX20 mutation, c.526G>A (p.D176N), was identified and co-segregated in all affected members in this family. This mutation was predicted to be deleterious by bioinformatics programs (SIFT, Polyphen2, and MutationTaster). This mutation was also not presented in the current Single Nucleotide Polymorphism Database (dbSNP) or National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP). In conclusion, our finding expands the spectrum of TBX20 mutations and provides additional support that TBX20 plays important roles in cardiac development. Our study also provided a new and cost-effective analysis strategy for the genetic study in small CHD pedigree.
| Family | Age | CHD | Echocardiography |
TBX20 |
||||
| ASD size (mm) | RA (mm) | RV (mm) | LVEF (%) | DNA | Protein | |||
| III1 (proband) | 7 months | ASD | 15 | 29 | 26 | 69 | 526G>A | D176N |
| I1 | 59 years | ASD | 2 | 35 | 34 | 60 | 526G>A | D176N |
| I2 | 61 years | No | 33 | 30 | 62 | |||
| II1 | 31 years | No | 32 | 30 | 65 | |||
| II2 | 28 years | ASD | 12 | 43 | 41 | 63 | 526G>A | D176N |
| II3 | 25 years | No | 32 | 31 | 69 | |||
| III2 | 3 years | No | 24 | 22 | 72 | |||

| Gene | Chr | Base position | RB | AB | Mutation | Amino acid alteration | Sorting intolerant from tolerant | Polyphen2 | MutationTaster |
| NOTCH2NL | chr1 | 145273345 | T | C | Missense | S>P | rs10910779 | ||
| NOTCH2 | chr1 | 120539661 | C | T | Missense | R>Q | rs146498360 | ||
| OBSCN | chr1 | 228562288 | G | A | Missense | G>R | Damaging (0.004) | PD (0.997) | DC (125) |
| USF1 | chr1 | 161011931 | T | G | Missense | Y>C | Tolerated (0.199) | PD (0.560) | DC (194) |
| ZNF638 | chr2 | 71576412 | A | G | Missense | I>V | rs12612365 | ||
| ZNF638 | chr2 | 71650308 | G | A | Missense | A>T | Damaging (0.024) | Benign (0.094) | Polymorphism (58) |
| VEGFC | chr4 | 177605086 | C | T | Missense | M>I | Tolerated (0.103) | Benign (0.000) | Polymorphism (10) |
| DST | chr6 | 56472194 | C | T | Missense | C>Y | rs185733722 | ||
| TBX20 | chr7 | 35288308 | C | T | Missense | D>N | Damaging (0.004) | PD (0.985) | DC (23) |
| LRRC6 | chr8 | 133634908 | G | T | Missense | P>H | rs76147813 | ||
| LDB3 | chr10 | 88469751 | C | T | Missense | A>V | Tolerated (0.291) | PD (0.745) | DC (64) |
| PTPN11 | chr12 | 112892433 | T | G | Nonsense | Y>* | rs76982592 | ||
| PTPN11 | chr12 | 112892407 | T | G | Missense | S>A | rs79068130 | ||
| MYH6 | chr14 | 23855762 | A | T | Missense | I>N | Damaging (0.000) | Benign (0.248) | DC (194) |
| MYH6 | chr14 | 23871682 | C | T | Missense | G>S | rs148962966 | ||
| IFT20 | chr17 | 26658963 | T | G | Missense | N>H | Damaging (0.015) | DC (68) | |
| DSC2 | chr18 | 28651796 | G | T | Missense | R>S | Tolerated (0.382) | Benign (0.095) | Polymorphism (110) |
| DOT1L | chr19 | 2211146 | T | C | Missense | V>A | Damaging (0.014) | Benign (0.001) | Polymorphism (64) |
| EP300 | chr22 | 41527628 | A | G | Missense | S>G | rs146242251 |
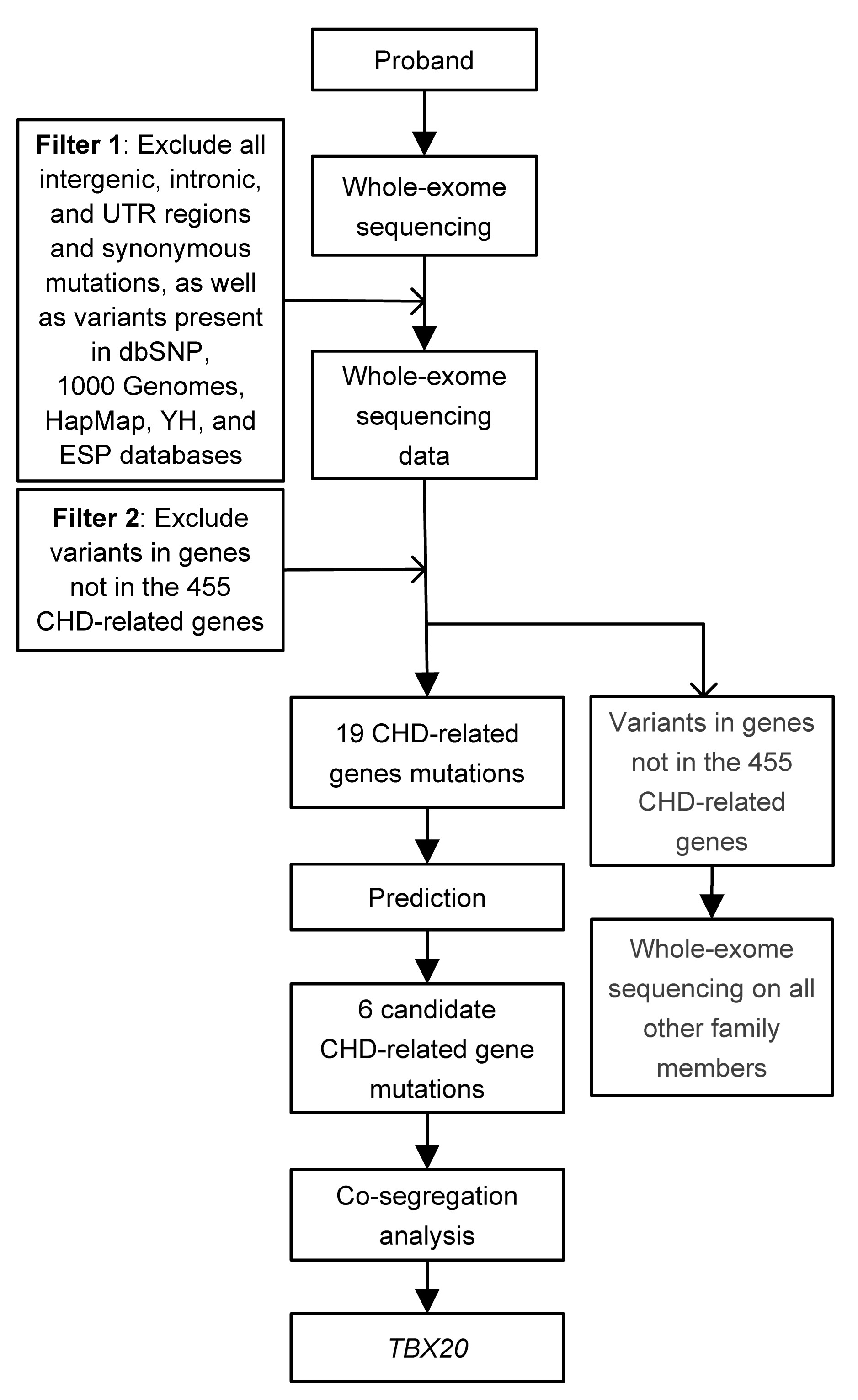
| Reference | Nucleotide change | AA change | Cardiac defect |
| Kirk et al. (2007) | 456C>G | I152M | ASD,VSD, PFO |
| 583C>T | Q195* | ASD, CoA, MVP, MR, DCM | |
| Liu et al. (2008) | 187G>A | A63T | ASD |
| 361A>T | I121F | TAPVC, ASD | |
| Qian et al. (2009) | 597C>G | H186D | ASD, MR, TOF, cleft mitral valve |
| 601T>C | L197P | ASD, TOF | |
| Posch et al. (2010) | 374C>G | I121M | ASD, TOF, cardiac valve defect |


Open peer comments: Debate/Discuss/Question/Opinion
<1>